Abstract
In this chapter, we introduce the concept of hemolytic anemia and classify them into corpuscular defects due to abnormalities in membrane, enzymes or hemoglobin and extracorpuscular defects due to immune, acquired and mechanical hemolytic anemias.
This chapter discusses common membrane defects and red cell enzymopathies. The membrane defects are presented with emphasis on the function of the membrane skeleton and the genetic and structural correlations between changes in red cell membrane proteins and clinical phenotypes. The pathogenesis, clinical manifestations, complications, diagnosis, and management of paroxysmal nocturnal hemoglobinuria is discussed. The chapter discusses pyruvate kinase deficiency and glucose-6-phosphate dehydrogenase deficiency and mentions other rare red cell enzymopathies.
Keywords
Hemolytic anemia, extravascular hemolysis, intravascular hemolysis, membrane skeletal defects, splenectomy, paroxysmal nocturnal hemoglobinuria (PNH), red cell enzymes, red cell membrane disorders
Hemolysis is a reduction in the normal red cell survival of 120 days. It may result from corpuscular abnormalities such as membrane, cytoskeleton, enzyme, or hemoglobin defects; or from extracorpuscular abnormalities involving immune or non immune mechanisms (see Chapter 10 ).
Clinical Features of Hemolytic Disease
The following clinical features suggest a hemolytic process in a child with anemia:
- 1.
History of anemia, jaundice, or gallstones in family.
- 2.
Persistent or recurrent anemia associated with reticulocytosis.
- 3.
Anemia unresponsive to hematinics.
- 4.
Intermittent bouts or persistent indirect hyperbilirubinemia/jaundice.
- 5.
Splenomegaly.
- 6.
Hemoglobinuria.
- 7.
Presence of multiple gallstones.
- 8.
Chronic leg ulcers.
- 9.
Development of anemia or hemoglobinuria after exposure to certain drugs.
- 10.
Cyanosis without cardiorespiratory distress.
- 11.
Polycythemia (2,3-diphosphoglycerate mutase deficiency).
- 12.
Dark urine due to dipyroluria (unstable hemoglobins, thalassemia, and ineffective erythropoiesis).
- 13.
Ethnic factors:
- a.
Incidence of sickle gene carrier in the African-American population (8%).
- b.
High incidence of thalassemia trait in people of Mediterranean ancestry.
- c.
High incidence of glucose-6-phosphate dehydrogenase (G6PD) deficiency among those with ethnic origins arising in territories near the Mediterranean Sea, Africa, and Southeast Asia.
- d.
High incidence of hereditary ovalocytosis in Southeast Asian populations.
- a.
- 14.
Age factors:
Anemia and jaundice in a Rh-positive infant born to a mother who is Rh negative or a group A or group B infant born to a group O mother (setting for a hemolytic anemia).
Laboratory Findings
Laboratory findings of hemolytic anemia consist of:
- •
Evidence of accelerated hemoglobin catabolism due to reduced red cell survival.
- •
Evidence of increased erythropoiesis.
Accelerated Hemoglobin Catabolism
Accelerated hemoglobin catabolism varies with the type of hemolysis as follows:
- •
Extravascular hemoglobin catabolism (see Figure 9.1 ).

Figure 9.1
Extravascular hemoglobin catabolism following extravascular hemolysis.
- •
Intravascular hemoglobin catabolism (see Figure 9.2 ).
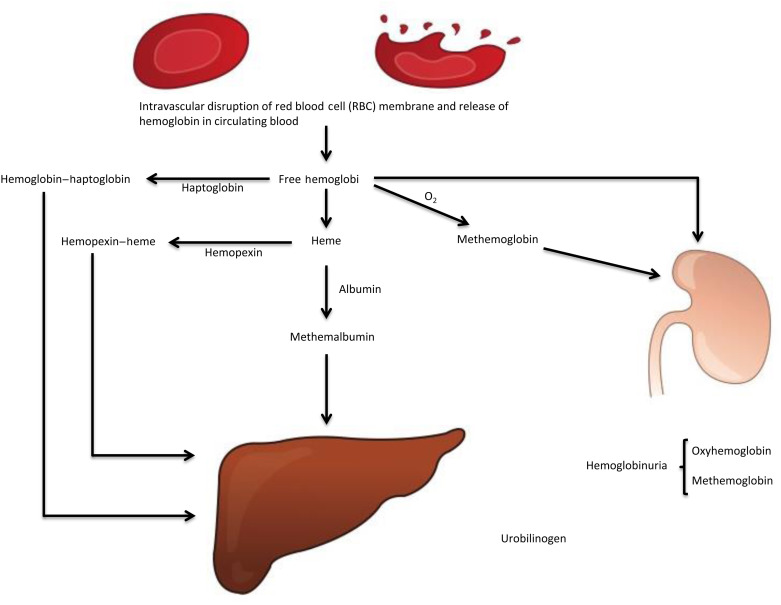
Figure 9.2
Intravascular hemoglobin catabolism following intravascular hemolysis. Hemoglobin–haptoglobin, hemopexin–heme, and methemalbumin are cleared by hepatocytes. Heme is converted to iron and bilirubin. The common pathway for both extravascular and intravascular hemolysis is the conjugation of bilirubin (bilirubin glucuronide) by the hepatocytes, its excretion in bile and ultimately formation of urobilinogen by the bacteria in the gut. Part of urobilinogen enters the enterohepatic circulation and part is excreted by the kidney in urine and the remainder of urobilinogen is excreted in stool.
The two may not be easily distinguished if the cause for hemolysis is not obvious, hence the long lists of markers of testing indicated below. The presence of hemoglobinuria and hemosiderinuria and the absence of haptoglobin are the major markers of intravascular hemolysis in practice.
Markers of Extravascular Hemolysis
- 1.
Increased unconjugated bilirubin.
- 2.
Increased lactic acid dehydrogenase in serum.
- 3.
Decreased plasma haptoglobin (normal level, 36–195 mg/dl).
- 4.
Increased fecal and urinary urobilinogen.
- 5.
Increased rate of carbon monoxide production.
Markers of Intravascular Hemolysis
- 1.
Increased unconjugated bilirubin (although often less than extravascular hemolysis as urinary losses leave less hemoglobin to be scavenged and processed to bilirubin).
- 2.
Increased lactic acid dehydrogenase in serum.
- 3.
Hemoglobinuria ( Figure 9.3 lists the causes of hemoglobinuria).

Figure 9.3
Causes of hemoglobinuria.
- 4.
Low or absent plasma haptoglobin.
- 5.
Hemosiderinuria (due to sloughing of iron-laden tubular cells into urine).
- 6.
Raised plasma hemoglobin level (normal value <1 mg hemoglobin/dl plasma, visibly red plasma contains greater than 50 mg hemoglobin/dl plasma).
- 7.
Raised plasma methemalbumin (albumin bound to heme; unlike haptoglobin, albumin does not bind intact hemoglobin).
- 8.
Raised plasma methemoglobin (oxidized free plasma hemoglobin) and raised levels of hemopexin–heme complex in plasma.
Increased Erythropoiesis
Erythropoiesis increases in response to a reduction in hemoglobin and is manifested by:
- 1.
Reticulocytosis : Frequently up to 10–20%; rarely, as high as 80%.
- 2.
Increased mean corpuscular volume (MCV) due to the presence of reticulocytosis.
- 3.
An increased red cell distribution width (RDW) as the hemoglobin level falls.
- 4.
Normoblasts in the peripheral blood.
- 5.
Specific morphologic abnormalities : Sickle cells, target cells, basophilic stippling, irregularly contracted cells or fragments (schistocytes), elliptocytes, acanthocytes, and spherocytes.
- 6.
Erythroid hyperplasia of the bone marrow : Erythroid:myeloid ratio in the marrow increasing from 1:5 to 1:1.
- 7.
Expansion of marrow space in chronic hemolysis resulting in:
- a.
Prominence of the frontal bones and broadened cheekbones.
- b.
Widened intratrabecular spaces with hair-on-end appearance of skull radiographs.
- c.
Biconcave vertebrae with fish-mouth intervertebral spaces.
- a.
- 8.
Decreased red cell survival demonstrated by 51Cr red cell labeling.
- 9.
Red cell creatine levels increased.
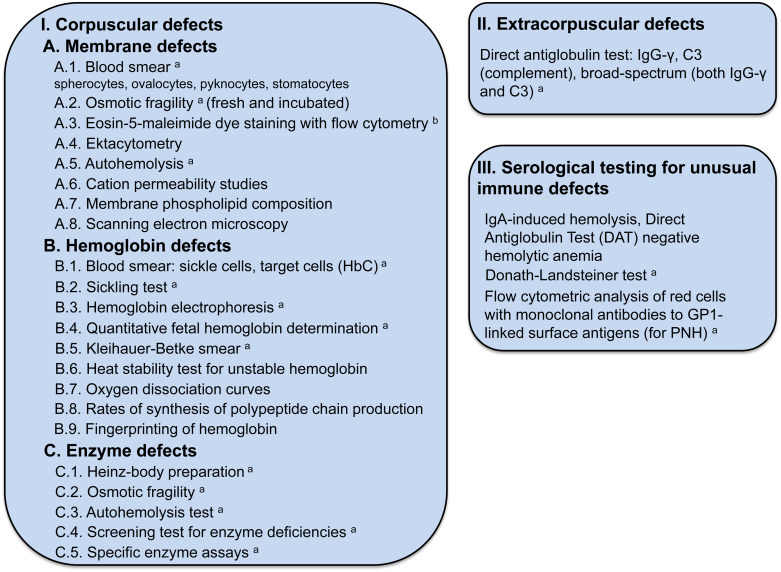
Figure 9.4 lists the tests used to establish the cause of the hemolytic anemia.
Membrane Defects
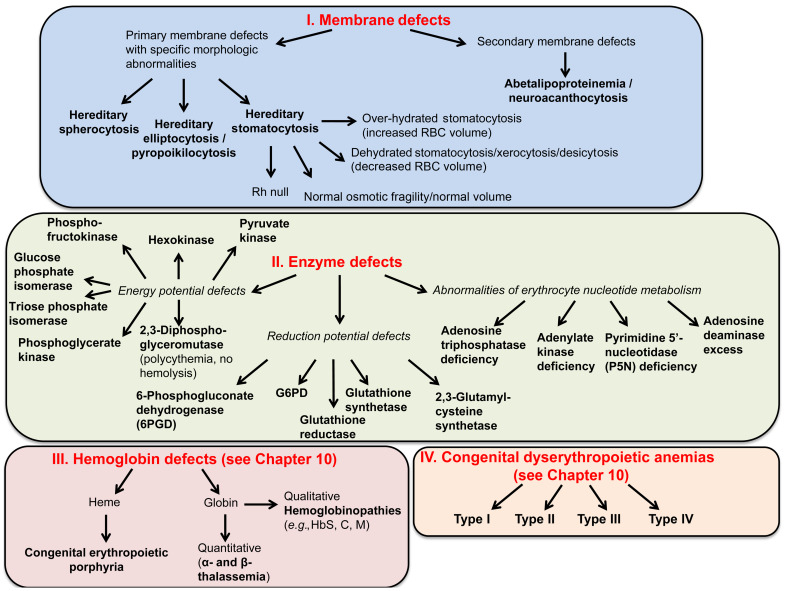
Figure 9.5 lists causes of hemolytic anemia due to corpuscular defects.
Structure of the Red Cell Membrane
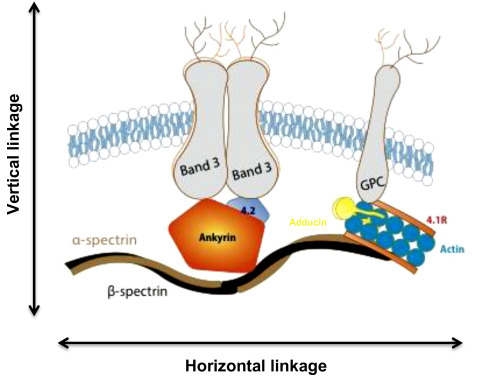
Spectrin, the major red cell membrane protein, is largely responsible for maintaining the normal red cell shape and overall morphology. It is composed of two large subunits, α- and β-spectrin, which are encoded by separate genes and are structurally distinct. Spectrin is integrated vertically into the lipid bilayer of the red cell membrane through the intercession of smaller proteins (ankyrin, 4.2) to integral membrane-spanning proteins (band 3, Rh antigen and glycophorin A). These vertical interactions maintain red cell membrane cohesion. Spectrin associates with itself head to head, while the tail associates with actin and other members of the junctional complex (4.1R, adducin). These horizontal interactions maintain membrane stability. Figure 9.6 summarizes the structure of the normal red cell membrane, highlighting the vertical and horizontal interactions.
The red cell membrane is semipermeable and must maintain its volume in order for the erythrocyte to negotiate the narrower spaces in the circulatory system. Red cell volume is maintained by a number of passive, gradient-driven cation and anion channels as well as active transporters.
Red Cell Membrane Disorders
Hereditary spherocytosis, elliptocytosis, stomatocytosis, acanthocytosis, xerocytosis, and pyropoikilocytosis can be diagnosed on the basis of their characteristic morphologic abnormalities.
Alterations in the quality and/or quantity of the proteins involved in the maintenance of the unique properties of the red cell membrane (deformability and stability) lead to the red cell membrane disorders:
- •
Hereditary spherocytosis (HS): perturbations in the vertical linkage.
- •
Hereditary elliptocytosis (HE): perturbations in the horizontal.
- •
Stomatocytosis: perturbations in the function of the ion transporters.
Note: Enzyme defects and many hemoglobinopathies have non-specific morphologic abnormalities related to secondary effects on red cell membrane proteins and pumps (e.g., ATP depletion).
Hereditary Spherocytosis
Genetics
- •
Autosomal dominant inheritance (75% of cases). The severity of anemia and the degree of spherocytosis may not be uniform within an affected family.
- •
No family history in 25% of cases. Some show minor laboratory abnormalities suggesting a carrier (recessive) state. Others are due to a de novo mutation.
- •
Most common in people of northern European heritage, with an incidence of 1 in 5000.
Pathogenesis
The primary defect is membrane instability due to dysfunction or deficiency of a red cell skeletal or membrane protein, including:
- •
Ankyrin mutations : Account for 50–67% of HS. In many patients, both spectrin and ankyrin proteins are deficient. Mutations of ankyrin occur in both dominant and recessive forms of HS. The clinical course varies from mild to severe. Red cells are typically spherocytes.
- •
α-Spectrin mutations occur in recessive HS and account for less than 5% of HS. Clinical course is severe. Contracted cells, poikilocytes, and spherocytes are seen.
- •
β-Spectrin mutations occur in dominant HS and account for 15–20% of HS. Clinical course is mild to moderate. Acanthocytes, spherocytic elliptocytes, and spherocytes are seen.
- •
Protein 4.2 mutations occur in the recessive form of HS and account for less than 5% of HS. Clinical course is mild to moderate. Spherocytes, acanthocytes, and ovalocytes are seen.
- •
Band 3 mutations occur in the dominant form of HS and account for 15–20% of HS. Clinical course is mild to moderate. Spherocytes are occasionally mushroom-shaped or pincered cells.
Deficiency of these proteins in HS results in a vertical defect, which causes progressive loss of membrane lipid and surface area. The loss of surface area results in characteristic microspherocytic morphology of HS red cells. The sequelae are as follows:
- •
Depletion of membrane lipid.
- •
Decrease in membrane surface area relative to volume, resulting in a decrease in surface area-to-volume ratio.
- •
Tendency to spherocytosis.
- •
Influx and efflux of sodium increased; cell dehydration.
- •
Sequestration of red cells in the spleen due to reduced erythrocyte deformability.
- •
Rapid adenosine triphosphate (ATP) utilization and increased glycolysis leading to increased loss of surface area under ATP-depleted conditions. This leads to the observation of splenic conditioning where the changes in glucose utilization, as well as cell volume control, are dramatically exacerbated with each circulatory passage through the spleen.
- •
Premature red cell destruction.
Hematologic Features
- 1.
Anemia : Mild to moderate when there is a compensated hemolytic anemia. In erythroblastopenic (aplastic or hypoplastic) crisis, hemoglobin may drop to 2–3 g/dl.
- 2.
MCV usually decreased; mean corpuscular hemoglobin concentration (MCHC) raised and RDW elevated.
- •
The MCHC is raised in HS, hereditary xerocytosis, hereditary pyropoikilocytosis (HPP), pyruvate kinase (PK) deficiency (which has acquired xerocytosis), and cold agglutinin disease. The presence of elevated RDW and MCHC (performed by aperture impedance instruments, e.g., Coulter) makes the likelihood of HS very high, because these two tests used together are very specific for HS.
- •
- 3.
Reticulocytosis (3–15%).
- 4.
Blood film : Spherocytes, microspherocytes (vary in number); hyperdense cells with or without polychromasia. The percentage of microspherocytes is the best indicator of the severity of the disease but not a good discriminator of the HS genotype. Hyperdense cells are seen in HbSC disease, HbCC disease, and xerocytosis. In HS, hyperdense cells are a poor indicator of disease severity but an effective discriminating feature of the HS phenotype.
- 5.
Direct antiglobulin test (DAT) negative.
- 6.
Increased red cell osmotic fragility (spherocytes lyse in higher concentrations of saline than normal red cells) occasionally only demonstrated after incubation of blood sample at 37 °C for 24 h (therefore, always do this test incubated). In spite of normal osmotic fragility, increased MCHC or an increase in hyperdense red cells is highly suggestive of HS.
- 7.
Autohemolysis at 24 and 48 h increased, corrected by the addition of glucose.
- 8.
Survival of 51 Cr-labeled cells reduced with increased splenic sequestration.
- 9.
Marrow: Normoblastic hyperplasia; increased iron.
- 10.
Eosin-5-maleimide dye staining of red cells and analysis by flow cytometry is the test of choice to diagnose HS but is only available in special reference laboratories.
- 11.
Genetic analysis for the alpha and beta spectrin, ankyrin, and band 3 mutations is available, but rarely necessary to be performed for diagnosis.
Biochemical Features
- 1.
Raised bilirubin, mainly indirect.
- 2.
Obstructive jaundice with increased direct bilirubin; may develop due to gallstones, a consequence of increased pigment excretion.
Clinical Features
- 1.
Anemia and jaundice: Severity depends on rate of hemolysis, degree of compensation of anemia by reticulocytosis and ability of liver to conjugate and excrete indirect hyperbilirubinemia.
- 2.
Splenomegaly.
- 3.
Presents in newborn period in 50% of cases with hyperbilirubinemia, reticulocytosis, normoblastosis, spherocytosis, negative DAT, and splenomegaly. Patients may present with a requirement for transfusion in the first 8 weeks of life that may not be reflective of their ultimate clinical severity.
- 4.
Presents before puberty in most patients.
- 5.
Diagnosis sometimes made much later in life, often after the birth of an infant with neonatal jaundice caused by HS.
- 6.
Coinheritance of HS with hemoglobin S-C disease may increase the risk of splenic sequestration crisis.
- 7.
Coinheritance of α- or β-thalassemia trait and HS has been reported to have variable effects on hemolysis.
- 8.
Iron deficiency may correct the laboratory values (artificially reducing the MCHC, etc.) but not the red cell lifespan in HS patients.
- 9.
HS with other system involvement:
- •
Interstitial deletion of chromosome 8p11.1–8p21.1 causes ankyrin deficiency, psychomotor retardation, and hypogonadism.
- •
HS may be associated with neurologic abnormalities such as cerebellar disturbances, muscle atrophy, and a tabes-like syndrome.
- •
In patients presenting with common bile duct obstruction associated with gallstones, the increased cholesterol and triglyceride load from the induced dyslipidemia can correct the membrane defect and the resulting spherocyte morphology, MCHC, and osmotic fragility results, hence masking the diagnostic features of the disease. Removal of bile duct obstruction leads to a reappearance of the disease phenotype.
- •
Classification
Table 9.1 lists a classification of HS in accordance with clinical severity and indications for splenectomy.
| Classification | Trait | Mild spherocytosis | Moderate spherocytosis | Severe spherocytosis a |
|---|---|---|---|---|
| Hemoglobin (g/dl) | Normal | 11–15 | 8–12 | 6–8 |
| Reticulocyte count (%) | ≤3 | 3.1–6 | ≥6 | ≥10 |
| Bilirubin (mg/dl) | ≤1.0 | 1.0–2.0 | ≥2.0 | ≥3.0 |
| Reticulocyte production index | <1.8 | 1.8–3 | >3 | |
| Spectrin per erythrocyte b (percentage of normal) | 100 | 80–100 | 50–80 | 40–60 |
| OSMOTIC FRAGILITY | ||||
| Fresh blood | Normal | Normal to slightly increased | Distinctly increased | Distinctly increased |
| Incubated blood | Slightly increased | Distinctly increased | Distinctly increased | Distinctly increased |
| AUTOHEMOLYSIS | ||||
| Without glucose (%) | >60 | >60 | 0–80 | 50 |
| With glucose (%) | <10 | ≥10 | ≥10 | ≥10 |
| Splenectomy | Not necessary | Usually not necessary during childhood and adolescence | Necessary during school age before puberty | Necessary, not before 5 years of age |
| Symptoms | None | None | Pallor, erythroblastopenic crises, splenomegaly, gallstones | Pallor, erythroblastopenic crises, splenomegaly, gallstones |
Diagnosis
- •
Clinical features and family history.
- •
Hematologic features.
Complications
- 1.
Hemolytic crisis : With more pronounced jaundice due to accelerated hemolysis (may be precipitated by viral infection).
- 2.
Erythroblastopenic crisis ( hypoplastic crisis ): Dramatic fall in hemoglobin level (and reticulocyte count); usually due to maturation arrest and often associated with giant pronormoblasts in the recovery phase; often associated with parvovirus B19 infection.
Parvovirus B19 infects developing normoblasts, causing a transient cessation of production. The virus specifically infects CFU-E and prevents their maturation. Giant pronormoblasts are seen in bone marrow. Diagnosis is made by increased IgM antibody titer against parvovirus and polymerase chain reaction for parvovirus on bone marrow.
- 3.
Folate deficiency : Caused by increased red cell turnover; may lead to superimposed megaloblastic anemia. Megaloblastic anemia may mask HS morphology as well as its diagnosis by osmotic fragility.
- 4.
Gallstones : In approximately one-half of untreated patients; increased incidence with age, can occur as early as 4–5 years of age. Occasionally, HS may be masked or improved in obstructive jaundice due to increase in surface area of red cells and the coinheritance of Gilbert syndrome markedly increases the incidence of gallstones.
- 5.
Complications of chronic anemia . Patients with more severe HS (see Table 9.1 ) may suffer growth retardation, anemic heart failure and failure to thrive, necessitating intermittent or chronic transfusion.
- 6.
Hemochromatosis: Rarely. This may occur more frequently when a restricted or partial splenectomy is carried out (see below).
- 7.
Splenic rupture: The risk of splenic rupture in HS is similar to that of the normal population. Nonetheless, a patient with a large spleen below the costal margin should be cautioned against contact sports or other activities known to lead to blunt trauma to the abdomen.
Treatment
- 1.
Folic acid supplement (1 mg/day).
- 2.
Leukocyte-depleted packed red cell transfusion for severe erythroblastopenic crisis.
- 3.
Splenectomy for moderate to severe cases (see Table 9.1 ). Most patients with less than 80% of normal spectrin content require splenectomy. Splenectomy should be carried out early in severe cases but not before 5 years of age, if possible. The management of the splenectomized patient is detailed in Chapter 4 . Although spherocytosis persists post splenectomy, the red cell lifespan normalizes and complications are prevented, especially transient erythroblastopenia and hyperbilirubinemia. There may, however, be an increased risk of arterial and venous thrombosis in later life as well as an increased risk for idiopathic pulmonary hypertension. Patients are at risk of sepsis after splenectomy, especially for those under 5 years of age. In partial splenectomy, up to 90% of the splenic mass is removed leaving enough splenic tissue to protect against infection. The technique is not widely utilized and its use should be primarily in transfusion-dependent patients who are under 5 years of age. There may be an increased risk for iron loading in patients with HS who have not undergone splenectomy.
Since patients in this situation are unlikely to tolerate phlebotomy, iron overload may make the decision for splenectomy or even partial splenectomy even more complex. Laparoscopic splenectomy is safe in children. Although it requires more operative time than open splenectomy, it is superior with regard to postoperative analgesia, smaller abdominal wall scars, duration of hospital stay and more rapid return to a regular diet and daily activities. It is not known whether accessory spleens are readily identified with the laparoscope although the magnification afforded by the laparoscope might be advantageous in some cases.
- 4.
Ultrasound should be carried out before splenectomy to exclude the presence of gallstones. If present, cholecystectomy is also indicated.
Hereditary Elliptocytosis
HE is clinically and genetically a heterogeneous disorder.
Genetics
HE is characterized by an autosomal dominant or codominant mode of inheritance with variable penetrance, affecting about 1 in 25,000 of the population. The prevalence of HE is much higher in regions where malaria is endemic. This could be explained by the resistance of elliptocytes to malarial invasion.
Occasionally, patients who are severely affected appear to be the offspring of a family with only a single affected parent. In this case a “silent carrier”-like mutation in an α-spectrin gene of the unaffected parent may be the cause.
Pathogenesis
HE is due to various defects in the skeletal proteins, spectrin, and protein 4.1, but also in the integral protein glycophorin C. The basic membrane defects consist of:
- •
Defects of spectrin self-association involving the α-chains (65%).
- •
Defects of spectrin self-association involving the β-chains (30%).
- •
Deficiency of protein 4.1.
- •
Deficiency of glycophorin C.
- •
“Silent carrier” effect: α-spectrin mutant genes which produce less α-spectrin when paired with an α-spectrin structural mutant. They lead to more severe disease (see below).
The mechanically unstable membrane of HE leads to shape change from discocyte to elliptocyte as the membrane is buffeted by sheer stress in the circulation.
Patients who are heterozygotes for these defects have milder disease while double heterozygotes and homozygotes for these mutants have progressively more severe syndromes.
Hematologic Features
- •
Blood smear: 25–90% of cells have elongated oval elliptocytes.
- •
Osmotic fragility is normal or increased.
- •
Autohemolysis is usually normal but may be increased and usually corrected by the addition of glucose or ATP.
Clinical Features
- •
Varies from patients who are symptom-free to severe anemia requiring blood transfusions. The percentage of microcytes best reflects the severity of the disease.
- •
About 12% have symptoms indistinguishable from HS.
- •
The percentage of elliptocytes varies from 50 to 90%. No correlation has been established between the degree of elliptocytosis and the severity of the anemia.
Classification
HE has been classified into the following clinical subtypes:
- •
Common HE, which is divided into several groups:
- a.
Silent carrier state.
- b.
Mild HE.
- c.
HE with infantile pyknocytosis.
- a.
- •
Common HE with chronic hemolysis, which is divided into two groups:
- a.
HE with dyserythropoiesis.
- b.
Homozygous common HE, which is clinically indistinguishable from HPP (see later discussion).
- a.
- •
Spherocytic HE, which clinically resembles HS; however, a family member usually has evidence of HE.
- •
Southeast Asian ovalocytosis, in which the majority of cells are oval. Some red cells contain either a longitudinal or transverse ridge.
- •
Infantile hemolytic elliptocytosis of infancy: These patients present with hemolytic elliptocytosis (mimicking HPP) which changes over the first 2 years of life to a clinical picture of mild HE as fetal hemoglobin changes to adult hemoglobin. Usually there is a single affected parent with HE.
Treatment
The indications and considerations for transfusion, splenectomy, and prophylactic folic acid are the same as for HS.
Hereditary Pyropoikilocytosis
Genetics
Homozygous or doubly heterozygous for spectrin chain mutants (e.g., Sp-a 1/74 and Sp-a 1/76 ). The spectrin chain defects found in HPP are similar to those found in HE.
Pathogenesis
HPP is a congenital hemolytic anemia associated with in vivo red cell fragmentation and marked in vitro fragmentation of red cells at 45 °C. Because of the similarities in the membrane defect in this condition and HE, it is viewed as a subtype of HE.
Biochemical and Biophysical Features
- 1.
Increased ratio of cholesterol to membrane protein.
- 2.
Decreased cell deformability.
Clinical Features
- 1.
Anemia characterized by extreme anisocytosis and poikilocytosis:
- a.
Red cell fragments, spherocytes, and budding red cells (the red cells are exquisitely sensitive to temperature and fragment after 10 min of incubation time at 45–46°C in vitro ; heating for 6 h at 37°C explains in vivo formation of fragmented red cells and chronic hemolysis).
- b.
Hemoglobin level reduced to 7–9 g/dl.
- c.
Marked reduction in MCV and elevated MCHC.
- a.
- 2.
Jaundice.
- 3.
Splenomegaly.
- 4.
Osmotic fragility and autohemolysis increased.
- 5.
Mild HE present in a parent or sibling.
Differential Diagnosis
Similar cells are seen in microangiopathic hemolytic anemias, after severe burns or oxidant stress and in PK deficiency.
Treatment
In infancy these patients require intermittent transfusion for hypoplastic crises. Patients respond well to splenectomy with a rise in hemoglobin to 12 g/dl. Following splenectomy, hemolysis is decreased but not totally eliminated.
Hereditary Stomatocytosis
Definition and Genetics
The stomatocyte has a linear slit-like area of central pallor rather than a circular area. When suspended in plasma, the cells assume a bowl-shaped form. This hereditary hemolytic anemia of variable severity is characterized by an autosomal dominant mode of inheritance. There are two forms of this inherited disorder related to failure to maintain normal red cell volume:
- 1.
Overhydrated stomatocytosis (previously referred to as “hereditary stomatocytosis”).
- 2.
Dehydrated stomatocytosis (previously referred to as “hereditary desicytosis or xerocytosis”). This is characterized by a relative paucity of stomatocytes with cells that appear very hyperchromic.
Etiology
The cells contain high Na + and low K + concentrations. The disorder is probably due to a membrane and protein defect. Although both forms share the relative increase in red cell sodium, overhydrated stomatocytosis is associated with an increase in red cell volume as the total cation content increases from unbridled sodium entry while dehydrated stomatocytosis has a reduced red cell volume as the potassium cation loss is not matched by sodium accumulation. The cells are abnormally rigid and poorly deformable, contributing to their rapid rate of destruction. There are many biochemical variants. The properties of the stomatocytosis syndromes are listed in Table 9.2 .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


