23 Gene Therapy for Gliomas
Malignant gliomas are aggressive neoplasms that cannot be completely resected and result in high mortality rates even with aggressive adjuvant therapy. Despite the evolution of treatment paradigms, less than 10% of patients with high-grade gliomas survive more than 5 years.1–3 Gliomas are infiltrative tumors within the brain parenchyma but they rarely metastasize to distant locations.4 Local inoculation of gene therapy vectors that exhibit tropism for the central nervous system (CNS) thus may be ideally suited for these tumors, and also avoids the systemic toxicity associated with current chemotherapeutic regiments.5 A burgeoning understanding of the molecular and genetic features of gliomas has produced novel targeted gene therapies. The combined failure of the current standard of care along with promising preclinical studies has resulted in hope for discovering a clinically effective glioma gene therapy.
Initial gene therapy clinical trials began in the 1990s using retrovirus as a vector to transfect glioma cells with the herpes simplex virus type 1 (HSV1) gene thymidine kinase (TK).4 HSV1-TK is an example of suicide gene therapy. The HSV1-TK protein phosphorylates peripherally administered ganciclovir (GCV) and, following further phosphorylation by the cellular kinase, a highly cytotoxic GCV-triphosphate (GCV-TP) is produced. This GCV-TP is highly cytotoxic as it (1) inhibits DNA polymerases, thus resulting in DNA damage and cell death; (2) is antiangiogenic; and (3) has a strong immunostimulatory effect.6 Since gene therapy was initially attempted using this approach, numerous strategies using both viral and nonviral vectors to deliver suicide, tumor suppressor, apoptotic, and toxic genes have been employed (Table 23.1). Viruses have also been utilized in direct oncolytic and immunogene therapy. The successful treatment of glioblastoma multiforme (GBM) will likely require a multifaceted approach (Fig. 23.1).
 Suicide Gene Therapy
Suicide Gene Therapy
Gene therapy for treatment of gliomas was pioneered by suicide gene-prodrug therapies such as the aforementioned GCV-TP. The first of these clinical trials employed a nonreplicating retrovirus to express HSV-TK. Histological studies of these early trials demonstrated low levels of transduction (< 5% of tumor cells) with improved survival confined only to the smallest tumors. The antitumor effect was hypothesized to result, at least in part, from a bystander effect mediated by the production of cytokines by mononuclear cells that infiltrate the tumor following HSV-TK therapy. The bystander effect was demonstrated to produce significant tumor cell death despite less than 10% transfection rates.7 A multicenter phase 3 study randomized 248 patients with newly diagnosed GBM to receive surgical resection and radiotherapy or surgical resection, radiotherapy, and retroviral delivered HSV-TK followed by peripherally administered GCV. Unfortunately, this study found no difference in survival, safety, or time to tumor progression.8
The initial failures of gene therapy trials demonstrate the importance of having both an effective vector and gene therapy target. The failure of retrovirus as a vector and HSV1-TK in initial clinical trials was likely secondary to low transduction rates (a nonreplicating virus was used, and producer cells likely were rejected shortly after implantation) and low catalytic activity of HSV1-TK on GCV.9 In a comparison of transduction rates, lacZ-expressing adenovirus (Ad) was found to have much higher transduction rates (11%) than the retrovirus (~4%) in a murine model of human glioma.10 In a head-to-head clinical trial comparing Ad to retrovirus delivery of HSV-TK, a significant increase in survival was found in patients treated with adenoviral HSV-TK (15 months) versus retroviral delivered HSV-TK (7.4 month).11 Subsequent clinical trials demonstrated the safety of HSV-TK adenoviral vectors in doses up to 2.0 × 1011 plaque-forming units (pfu, active viral particles). Above this dose, patients had severe adverse reactions, including hyponatremia, altered mental status, increased intracranial pressure, and seizure activity. Although many of these clinical trials resulted in the occasional long-term survivor and evidence of increased long-term survival, no phase 3 trial has definitively shown a significant improvement in survival and tumor progression.4,12–15 The majority of side effects were due to inflammation and malignant edema. Inflammation produced by Ad particles was at least partially responsible for the tumor response seen in these trials.15
Pitfalls
• Suicide gene therapy appears to require transduction rates to be effective, but those rates have not been achieved in vivo.
• Gene catalytic activity on prodrugs requires further optimization.
• Certain preclinical models (e.g., C6 glioma) may result in artificially high antitumor immune responses.
• More than 15 suicide gene therapy trials (phases 1 to 3) have demonstrated no definitive increase in median survival.
Table 23.1 Overview of Gene Therapies
| Strategy | Examples of Genes | Mechanism |
| Suicide gene | HSV-TK, cytosine deaminase | Gene encodes an enzyme that converts a prodrug into a toxin |
| Restoring apoptotic pathways | p53, retinoblastoma p16, phosphatase and tensin homologue (PTEN) | Corrects mutations in apoptotic pathways in tumor cells |
| Immunotherapy | GM-CSF, TNF-α, interleukins, interferons, B-7, sFlt-3-L, ICAM | Enhanced presentation of tumor antigens, and cytokines resulting in activation of tumor-killing immune cells |
Abbreviations: HSV-TK, herpes simplex virus thymidine kinase; TNF, tumor necrosis factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICAM, intercellular adhesion molecule.
The HSV1-TK gene used in previous human trials required a very high dose of GCV to be administered. The near-immunosuppressive doses of GCV may have offset some of the immunostimulatory effects of the HSV1-TK suicide gene. The mutant gene HSV1-sr39TK exerts a 14-fold higher catalytic response than HSV1-TK, allowing for lower dosing of GCV. Preclinical studies in a C6 glioma model have demonstrated a stronger therapeutic efficacy. Further refinement of both the viral vector and gene may yield promising results in future clinical trials.
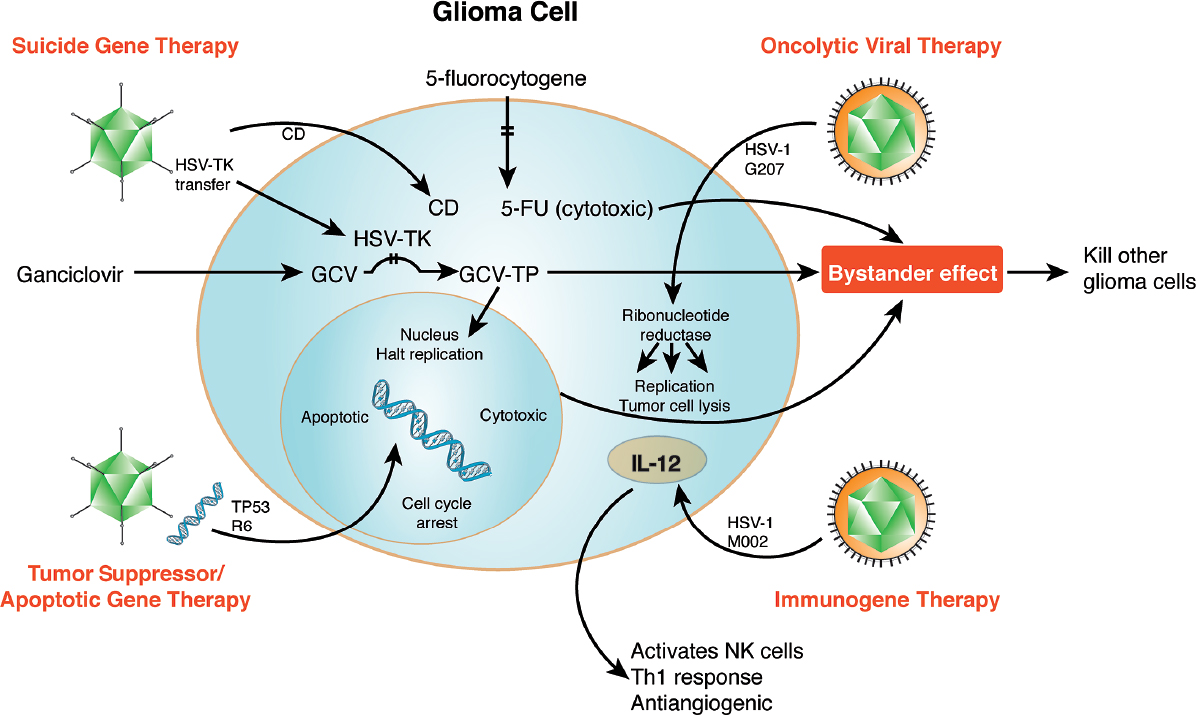
Fig. 23.1 Viral gene therapeutic approaches for gliomas fall within four categories. Suicide gene therapy (top left) involves HSV-TK converting peripherally administered GCV into cytotoxic GCV-TP. Tumor suppressor/apoptotic gene therapy (bottom left) can insert suppressor genes such as TP53 into the nucleus. This results in cell-cycle arrest and even apoptosis. Oncolytic viral therapy using HSV-1 G207 (top right) leads to tumor cell lysis through viral replica tion, enabled by the presence of cellular ribonucleotide reductase present in trans. Immunogene (bottom right) therapy produces an inflammatory response through interleukins (e.g., IL-12) that can result in both cell death and, in the case of IL-12, a decrease in angiogenesis. Additionally, peripheral administered medications (prodrugs) (top center) can be metabolized by microbial enzymes introduced into the cell through gene therapy, allowing the local (rather than systemic) creation of a cytotoxic intermediate (5-fluorouracil [5-FU]). This schematic illustrates multiple targets within glioma cells that can be synergistically attacked to result in local and bystander cell death.
Cytosine deaminase (CD)-5FC is another example of a suicide gene therapy. CD is not found in mammalian cells but is expressed in certain bacteria and fungi. The enzyme has been shown to deaminate 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU), an antimetabolite that interrupts RNA and DNA synthesis. This approach may have some advantages over the HSV-TK approach because fewer cells must be transfected, as 5-FU is a small molecule that passively diffuses between cells and has enhanced cytotoxic effect on surrounding cells (bystander effect). In comparison, GCV-TP is a large molecule and is dependent on connexins for transport to adjacent cells.16 The CD model has demonstrated effectiveness in preclinical glioma models, and a clinical trial is currently underway (clinicaltrials.gov, trial NCT01156584).17 The vector under study, Toca 511, has important advantages, utilizing a retroviral replicating vector encoding an optimized CD, and has demonstrated a threefold prodrug catalyzation in infected cells.18 Data available for the first three patients have not demonstrated any adverse reactions.17
 Tumor Suppressor and Apoptotic Gene Therapy
Tumor Suppressor and Apoptotic Gene Therapy
Glioblastoma multiforme has undergone the most extensive genetic profiling of any cancer, and therapies aimed at specific mutations may result in clinically significant improvements in survival.19,20 TP53, RB1, NF1, and PTEN are tumor suppressor genes implicated in GBM. Various vectors have been used in an attempt to repair these oncogenic mutations. These repairs are aimed at restoring apoptotic pathways and intrinsic cell-cycle regulation. Mutations of the TP53 gene are seen in 30 to 40% of GBM patients and the TP53 pathway is involved in upwards of 80% of tumors. As a result, the TP53 gene and pathway have been a primary target in attempts to restore normal cell-cycle regulation. A phase 1 clinical trial testing an Ad-p53 vector in 15 patients did not show significant toxicity but demonstrated transduction in only a small portion of the tumors. No significant increase in survival was noted.21 Another study of 18 patients demonstrated improvements in both survival and 6-month Karnofsky Performance Scale scores in the treatment group compared to the surgery-alone group (n = 20).22
Adenoviral vectors have also been used to transfer the Rb tumor suppressor gene to malignant gliomas. In vitro studies were promising, but transfection of tumor cells with Rb during in vivo studies was ineffective.23–25 The heterogeneity of genetic mutations in gliomas will likely restrict the utility of restoration therapy.26
 Nonviral Vectors
Nonviral Vectors
Nonviral vectors could potentially be a safer method of delivering genetic material than viral vectors as they are nonpathogenic and nonimmunogenic.27,28 In an early gene therapy trial, a patient with partial ornithine transcarbamylase deficiency died when the adenoviral vector spread beyond the liver, producing a vigorous immune response and multiple system organ failure.29 Concerns over the oncogenic potential of some viral vectors, recombination of viral vectors to virulent forms, and the insertion of viral genes into gametes have been raised.27 However, to date few adverse events and no deaths have occurred in glioma gene therapy trials as a result of viral vectors.8,15,21,30
Nonviral vectors range from naked DNA to complex chemical carriers to stem cells. The delivery of naked DNA into cells has extremely low transduction efficiency, but certain mechanical methods may increase gene uptake. These techniques include electroporation, gene gun delivery, ultrasound, and hydrodynamic (high-pressure) injection. Mechanical augmentation has improved gene transfer in systemic organs but is not yet practical for CNS use with the exception of convection-enhanced delivery.
The ability to customize cationic lipids and polymers for delivery of genes to specific tissues has dramatically improved the efficiency of this approach. Lipoplexes are DNA-liposomal complexes that have been widely used for gene delivery. Nonviral vectors have met with moderate success in glioma animal models. One study delivered an antisense RNA molecule directed at the epidermal growth factor receptor (EGFR).28 To overcome the blood–brain barrier, injections of bradykinin were given with the vector. To further increase efficiency of transfection, two monoclonal antibodies were bound to the vehicle: one for human insulin receptor and the other for murine transferrin receptor. This treatment resulted in a 95% suppression of EGFR function and an 88% increase in the survival times.28 Nonviral vectors have been implemented in immunotherapy for glioma as well. Interferon (IFN)-β delivered with a cationic liposome by stereotactic injection into an intracranial mouse model produced a significant increase in survival time and histologically produced an increased T-cell lymphocyte infiltrate. Of the mice receiving IFN-β gene therapy, 40 to 50% had complete cures, and upon rechallenge did not develop tumors over a 50-day period.31
The relatively low transfection rates of nonviral vectors and the susceptibility of plasmids to degradation led to the development of a Sleeping Beauty transposon-based vector to insert genes into nuclear DNA in the tumor cell, potentially resulting in long-term expression of the gene product, even after tumor cell mitosis. Such stable long-term production of protein would be ideal for antiangiogenic therapy. The failure of endostatin and other antiangiogenic trials was thought to be due to the relatively transient effect of antiangiogenic therapies. Two vectors were produced that delivered genes for soluble endothelial growth factor receptor (sFlt-1) and angiostatin-endostatin (statin-AE) fusion protein.32 Long-term production of these proteins was achieved by combining them with the Sleeping Beauty transposable element, and delivery was augmented by convection-enhanced delivery. sFlt-1 and statin-AE combined produced a significant increase in survival compared to controls and either gene alone.32,33
Pearl
• Nonviral vectors may avoid the antiviral immune response and potential toxicity of viral vectors.
• Although nonviral vector technology has potential advantages, transfection rates remain well short of those achieved by viral vectors.
Nonviral vectors require the manufacture of a vehicle that can attach to a target cell’s surface, be internalized, escape from endosomes, enter the nucleus, and initiate transcription.27 The ability to engineer chemical carriers that can more efficiently accomplish these goals has been remarkable and promising.34 However, at present nonviral vectors are 1,000 to 50,000 times less efficient at transfecting gliomas when compared with adenoviral vectors.27 This low transfection rate has prevented the emergence of a successful nonviral, acellular vector for clinical use.
Nonviral Cellular Vectors: Neural and Mesenchymal Stem Cells
Recent work suggests that stem cells are responsible for driving continued cell growth in many cancers, including GBM. These stem cells (also called glioma progenitor cells or glioma-initiating cells) are thought to constitute less than 5% of the tumor, are resistant to traditional therapy, and thus are largely responsible for recurrence.
Stem cells not only may be responsible for tumorigenesis but also may provide an avenue for treatment. Neural and mesenchymal stem cells have been used as gene therapy vectors. The promise of stem cells in treating gliomas was first realized when it was demonstrated that they exhibit tropism for sites of CNS injury. It was then demonstrated that neural stem cells exhibit tropism for gliomas.5 Obtaining neural stem cells can be difficult, and so mesenchymal stem cells, which are easily collected from bone marrow or adipose tissue, have been extensively studied. Several investigators have demonstrated that mesenchymal stem cells also localize to gliomas.35 The implications of selective tropism for gliomas enable these cellular vectors to potentially deliver a wide variety of therapies including the aforementioned gene therapies, IFN-β, secretable tumor necrosis factor-related apoptosis-inducing ligand (S-TRAIL), microRNA, and oncolytic viruses.35 Stem cells can potentially target the infiltrating tumor margins that have been resistant to traditional therapies.
The tumor selective tropism of bone-marrow–derived human mesenchymal stem cells (BM-hMSCs) for gliomas was recently found to be associated with transforming growth factor-β (TGF-β).36 BM-hMSCs have TGF-β receptors that respond to TGF-β secreted from glioma cells. BM-hMSCs also exhibit tropism for glioma stem cells that, as mentioned previously, are likely responsible for the majority of treatment failures and recurrences.36 Finally, BM-hMSC were shown to be an effective in vivo vector for the oncolytic Delta-24-RGD Ad when intravascularly delivered.
Mesenchymal stem cells enable the study of novel gene therapies that require a vector. For example, micro-RNAs (miRNAs) are small noncoding RNAs that can regulate proliferation, apoptosis, cell-cycle regulation, invasion, glioma stem cell function, and angiogenesis by augmenting or downregulating gene expression.37 Certain miRNAs appear to be attractive targets for glioma gene therapy.
Oncolytic Viral Therapy
Replication-incompetent viral vectors are inefficient and have not produced clinically significant improvements in survival during clinical trials to date. As a result, conditionally replicating viral vectors have been engineered to selectively replicate in tumor cells. These vectors can kill tumor cells from direct tumor cell lysis and from amplification of therapeutic genes (Table 23.2). Oncolytic viruses currently being studied in antiglioma clinical trials include oncolytic herpes simplex virus (oHSV), conditionally replicating Ad (CRAd), reovirus, poliovirus, vaccinia virus, measles virus, and Newcastle virus.38
Herpes Simplex Virus
A genetically engineered HSV-1 that conditionally replicated in tumor cells was created using a virus constructed with a deletion of the thymidine kinase (tk) gene, and it was effective in animal glioma models. This virus was never taken to human trials because the deletion of the tk gene rendered the virus resistant to acyclovir.39 The HSV-1 G207 was constructed, containing two mutations initially created in two separate viral constructs. The two mutations were combined in anticipation of clinical trials to prevent a potential in-situ recombination event from restoring a wild-type phenotype. G207 contains the deletions present in the γ1 34.5 region and a disabling lacZ insertion at the UL39 locus. UL39 encodes the large subunit of the viral ribonucleotide reductase that is necessary for the synthesis of nucleotides in postmitotic cells.40 Ribonucleotide reductase is necessary for viral replication and is present in trans in actively dividing cells. γ1 34.5-deleted HSVs replicate at lower levels than wild-type viruses, but these replication deficits can be significantly overcome by the administration of ionizing radiation shortly after virus inoculation.41,42 Radiation increases late viral protein production via activation of p38l; this increases viral replication and spread of infection. The increase in replication does not appear to result in an increase in toxicity.
Table 23.2 Overview of Viral Vectors
| Virus | Replicating Ability |




