FETAL COMPARTMENT
The understanding and elucidation of the human fetal endocrine system have required the development of assays for minute quantities of hormone. The regulation of the fetal endocrine system, like that of the placenta, is not completely independent, since synthesis relies to some extent on precursor hormones secreted directly by the placenta or obtained from the maternal unit. As the fetus develops, its endocrine system becomes more independent in preparation for extrauterine existence.
HYPOTHALAMIC-PITUITARY AXIS
The fetal hypothalamus begins differentiation from the forebrain during the first few weeks of fetal life, and by 12 weeks, hypothalamic development is well advanced. Most of the hypothalamic-releasing hormones, including GnRH, TRH, dopamine, norepinephrine, and somatostatin, and their respective hypothalamic nuclei have been identified as early as 6 to 8 weeks of fetal life.23 The neurohypophysis is detected first at 5 weeks, and by 14 weeks, the supraoptic and paraventricular nuclei are fully developed.23,51
Rathke pouch appears in the human fetus at 4 weeks. The premature anterior pituitary cells that develop from the cells lining Rathke pouch can secrete GH, prolactin, FSH, LH, and ACTH in vitro as early as 7 weeks of fetal life.52 Evidence suggests that the intermediate lobe of the pituitary is a significant source of POMC hormones.53,54
The hypothalamic-pituitary portal system is the functional link between the hypothalamus and the anterior pituitary. Vascularization of the anterior pituitary begins by 13 weeks of fetal life, but a functioning intact portal system is absent until 18 to 20 weeks.55 However, there is indirect evidence that hypothalamic secretion of releasing hormones influences anterior pituitary function before this time by simple diffusion, given their proximity in early fetal development. There is also evidence that fetal adrenal feedback is operative at the hypothalamic-pituitary axis as early as week 14 of fetal life. Elevated levels of fetal androgens are detected at this time in amniotic fluid in fetuses affected with congenital adrenal hyperplasia secondary to 21-hydroxylase deficiency.56
GROWTH HORMONE, PROLACTIN, VASOPRESSIN, AND OXYTOCIN
GH is detected in the fetal pituitary as early as 12 weeks’ gestation, and fetal pituitary GH concentrations increase until 25 to 30 weeks’ gestation, thereafter remaining constant until term. Fetal plasma GH levels peak at 20 weeks and then fall rapidly until birth57 (Fig. 108-4). However, fetal plasma concentrations of GH always exceed maternal concentrations, which are suppressed, possibly by the high circulating levels of hPL.
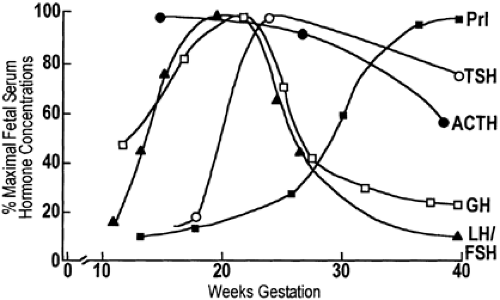 FIGURE 108-4. Ontogeny of pituitary hormones in human fetal serum. (Prl, prolactin; TSH, thyroid-stimulating hormone; ACTH, adrenocorticotropic hormone; GH, growth hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone.) (From Parker CR Jr. The endocrinology of pregnancy. In: Carr BR, Blackwell RE, eds. Textbook of reproductive medicine. Norwalk, CT: Appleton & Lange, 1993:17.) |
The regulation of GH release in the fetus appears to be more complex than in the adult. To explain the high levels of plasma GH at midgestation and a fall thereafter, unrestrained release of growth hormone–releasing hormone (GHRH) leading to excessive release of GH at midgestation has been postulated.23 Thereafter, as the hypothalamus matures, somatostatin may increase and GHRH levels decline, reducing GH release. The role of GH in the fetus is also unclear. There is considerable evidence that GH is not essential to somatic growth in primates.58 For example, in neonates with pituitary agenesis, congenital hypothalamic hypopituitarism, or familial GH deficiency, birth size and length are usually normal. However, the somatomedins, in particular IGF-I and IGF-II, increase in fetal plasma, and IGF-I and IGF-II levels correlate better than do GH levels with fetal growth.59 Although GH is an important trophic hormone for somatomedin production in the fetus, somatomedin regulation may be independent of GH, depending instead on other factors. Prolactin, hPL, and insulin also stimulate somatomedin production.23
Prolactin is present in lactotropes by 19 weeks of life. The prolactin content of the fetal pituitary increases throughout gestation,60 whereas plasma levels increase slowly until 30 weeks’ gestation, when the levels rise sharply until term and remain elevated until the third month of postnatal life (see Fig. 108-4). In humans, TRH and dopamine as well as estrogens appear to affect fetal prolactin secretion. Bromocriptine both lowers maternal prolactin levels and crosses the placenta to inhibit
fetal prolactin release and lower prolactin levels in fetal blood.61 It has been suggested that prolactin in the fetus influences adrenal growth, lung maturation, and amniotic fluid volume.
fetal prolactin release and lower prolactin levels in fetal blood.61 It has been suggested that prolactin in the fetus influences adrenal growth, lung maturation, and amniotic fluid volume.
Arginine vasopressin (AVP) and oxytocin are found in hypothalamic nuclei and in the neurohypophysis during early fetal development.52 However, there are relatively few studies on the regulation and secretion of these hormones. The levels of AVP are high in fetal plasma and cord blood at delivery. The principal stimulus to AVP release appears to be fetal hypoxia, although acidosis, hypercarbia, and hypotension also play a role.62 The elevated AVP levels in fetal blood may lead to increased blood pressure, vasoconstriction, and the passage of meconium by the fetus. Oxytocin levels in the fetus are not affected by hypoxia but appear to increase during labor and delivery (see Chap. 109).
THYROID GLAND
The placenta apparently is relatively impermeable to TSH and thyroid hormone, so that the fetal hypothalamic–pituitary– thyroid axis develops and functions independently of the maternal system. Although thyroxine (T4) may cross the placenta to a slight degree, human thyroid-stimulating immunoglobulins (TSI) as well as iodine and antithyroid drugs given to women with hyperthyroidism pass through the placenta and may affect fetal thyroid function.63
TRH is detectable in hypothalamic nuclei, and TSH is found in pituitary tissues by 10 to 12 weeks of fetal life.64 High concentrations of TRH are detected in fetal blood, and the source is thought to be the fetal pancreas. However, this source of TRH appears to have little effect on the pituitary release of TSH, and the function of pancreatic TRH is unknown.65
The fetal thyroid has developed sufficiently by the end of the first trimester that it can concentrate iodine and synthesize iodothyronines. The levels of TSH and thyroid hormone are relatively low in fetal blood until midgestation. At 24 to 28 weeks’ gestation, serum TSH concentrations rise abruptly to a peak but decrease slightly thereafter until delivery.66 In response to the surge of TSH, T4 levels rise progressively after midgestation until term (Fig. 108-5). During this time, both thyroid responsiveness to TRH and pituitary TRH content increase. At birth, there is an abrupt release of TSH, T4, and triiodothyronine (T3), and the levels of these hormones fall during the first few weeks after birth.66 The relative hyperthyroid state of the fetus is believed to be necessary to prepare it for the thermoregulatory adjustments of extrauterine life. The abrupt changes of TSH and T4 that occur at birth are believed to be stimulated by the cooling associated with delivery.67 Finally, 3,3′,5′-triiodo-L- thyronine (reverse T3) levels are high during early fetal life, begin to fall at midgestation, and continue to fall after birth (see Chap. 47). The difference between the formation of T3 and reverse T3 is thought to be related to maturation of peripheral iodothyronine metabolism (see Chap. 30).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


