Chapter Outline
Chromosomal Rearrangements in Ewing Sarcoma
EWS/FLI and Other TET/ETS Fusions in Ewing Sarcoma
Cooperative Pathways in Ewing Sarcoma Oncogenesis
Apoptotic Pathways in Ewing Sarcoma
CLINICAL PRESENTATION AND DIAGNOSIS
TREATMENT OF PATIENTS WITH NONMETASTATIC EWING SARCOMA
Systemic Treatment with Chemotherapy
Management of Tumors Arising at Special Sites
Summary of Outcomes for Patients with Nonmetastatic Ewing Sarcoma
TREATMENT OF PATIENTS WITH METASTATIC EWING SARCOMA
Role of Surgery and Radiation in Patients with Metastatic Disease
Role of High-Dose Therapy with Hematopoietic Stem Cell Rescue for Metastatic Disease
Summary of Outcomes for Patients with Metastatic Ewing Sarcoma
Location and Timing of Disease Recurrence
General Approach to Patients with Recurrent Ewing Sarcoma
Systemic Therapies for Recurrent Ewing Sarcoma
Role of Surgery and Radiation for Recurrent Ewing Sarcoma
Role of High-Dose Therapy with Hematopoietic Stem Cell Rescue for Recurrent Disease
LATE EFFECTS IN PATIENTS TREATED FOR EWING SARCOMA
In 1921 James Ewing described the cancer that came to carry his name: a primary bone tumor composed of small round blue cells and devoid of the malignant osteoid that characterizes osteosarcoma. Subsequently, pathologists described other clinicopathologic entities initially thought to be distinct, such as peripheral primitive neuroectodermal tumor (PNET) of bone or soft tissue or the Askin tumor of the chest wall. However, biologic study and clinical responses to chemotherapy have now linked these tumors into one entity or family of tumors, a grouping that also includes extraosseous Ewing sarcoma. Commonly used terms for this tumor include Ewing sarcoma, Ewing tumors, and Ewing sarcoma family of tumors. In this chapter the World Health Organization–approved term, Ewing sarcoma, is generally used. When appropriate, specific characteristics that differentiate Ewing sarcoma, PNET, and Askin tumors are described.
The chapter begins with a discussion of the epidemiology of Ewing sarcoma. Next, the current understanding of the cellular and molecular features of these tumors is reviewed. An overview of the clinical presentation and prognostic features of patients with this disease are presented. This overview is followed by a discussion of the management of patients with Ewing sarcoma. The chapter concludes with a review of the late effects seen in patients treated for these tumors.
Epidemiology
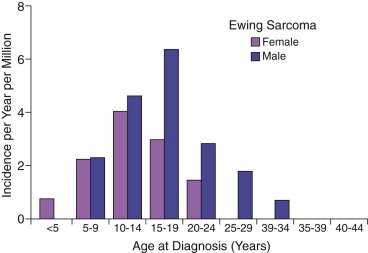
Ewing sarcoma is the second most common type of primary bone cancer in the United States and Europe, accounting for approximately 25% to 34% of malignant bone tumors. Approximately 250 new cases of Ewing sarcoma are diagnosed in the United States each year. In the United States Ewing sarcoma has had a relatively stable average incidence of 2.5 to 3 cases per million per year from 1975 to 2000. A similar age-adjusted incidence has been noted in Europe. As shown in Figure 61-1 , the peak incidence occurs during adolescence, with an average incidence of 4.6 cases per million per year for the 15- to 19-year age range. Younger children are affected less frequently, and the disease is distinctly uncommon in adults older than 35 years of age. A male predominance has been noted, with a male-to-female ratio of approximately 1.25 : 1. However, one study reported a female predominance in patients younger than 3 years of age at initial diagnosis. There is no seasonal variation in the appearance of this disease.
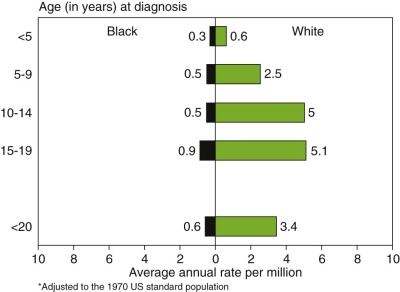
The incidence of Ewing sarcoma varies markedly among races and countries, with the highest frequency reported from Australia and Sao Paolo, Brazil. People of African ancestry appear to have the lowest incidence of this disease. Figure 61-2 demonstrates the dramatically lower incidence of these tumors in the U.S. African-American/Black population. A similar low incidence in sub-Saharan Africa has been reported and suggests a possible genetic difference in the risk of developing Ewing sarcoma. Patients of African ancestry with Ewing sarcoma have higher rates of soft tissue tumors and an older age distribution compared with patients of European ancestry, indicating that racial differences in this disease extend beyond differences in incidence. Ewing sarcoma also appears to be uncommon in people of East Asian ancestry, based on data from China, Japan, and Thailand. Data from the Middle East, North Africa, and the Indian subcontinent suggest that Ewing sarcoma is at least as common in these populations as in people of European ancestry. In small studies from Kuwait and Bombay, for example, Ewing sarcoma was identified as the most common primary malignant bone tumor.
Based on these geographic differences in the incidence of Ewing sarcoma, several groups have investigated the genetic epidemiology of this disease. One group reported differences between African and European populations in the number of Alu repeat sequences in intron 6 of the EWSR1 gene, upstream of the breakpoint region involved in translocations at this locus seen in most cases of Ewing sarcoma. Two studies further focused on this possibility that differences in the incidence of Ewing sarcoma may be due to differences in polymorphisms in the EWSR1 locus that could result in different rates of translocation. Both studies each identified a different single nucleotide polymorphism with low-level statistical association with Ewing sarcoma, although the effect of each polymorphism on the risk of Ewing sarcoma was small. Another study investigated the incidence of other tumors that harbor EWSR1 translocations across racial groups and observed higher rates of desmoplastic small round cell tumor in the U.S. African-American population compared with the U.S. European-American population. This finding of higher rates of an EWSR1 -associated tumor in a population with a lower rate of Ewing sarcoma provides additional evidence that differential susceptibility to translocation at the EWSR1 locus does not account for racial differences in the incidence of Ewing sarcoma. More recently, a genome-wide association study identified and validated polymorphisms in three loci, 1p36, 10q21, and 15q15, associated with increased risk of Ewing sarcoma. These risk variants are present at lower frequencies in African populations, providing one potential explanation for the lower rates of Ewing sarcoma in this population.
The etiology of Ewing sarcoma remains largely obscure. These tumors are not generally believed to be familial, although rare cases of sibling pairs with Ewing sarcoma have been reported. First- and second-degree relatives of patients with Ewing sarcoma do not appear to have an increased overall risk of cancer, although a higher rate of stomach cancer, melanoma, renal cancer, and brain tumors has been reported.
Patients with Ewing sarcoma appear to have an increased incidence of specific congenital anomalies. The most consistent finding has been an association between Ewing sarcoma and congenital hernias, particularly inguinal hernias. A pooled analysis of the available literature has confirmed this association, with an odds ratio of developing Ewing sarcoma of 2.8 for children with congenital hernias compared with children without congenital hernias. Whether a common hormonal or environmental mechanism underlies the development of both congenital hernias and Ewing sarcoma remains unclear. Patients with Ewing sarcoma have also been noted to have an increased incidence of bone anomalies, particularly rib and vertebral anomalies. Other congenital anomalies, including cataracts and genitourinary anomalies, have been reported in patients with Ewing sarcoma. These anomalies occur at a low incidence in these patients and without a consistent pattern, suggesting that their occurrence with Ewing sarcoma may be coincidental.
Various parental exposures have been associated with the risk of developing Ewing sarcoma. The most consistent finding has been an association between parental farm exposure around the time of conception through birth and the subsequent development of Ewing sarcoma. This association has been reported in a series of case-control studies and affirmed in a meta-analysis, with an increased risk if either parent was involved in farming. Specific exposures related to agricultural work that have been suggested as driving this increased risk include parental fertilizer, pesticide, solvent, and wood dust exposure. Less consistently reported associations include an increased risk of Ewing sarcoma with parental cigarette smoking and parental occupation in manual labor. Family income does not appear to be associated with the risk of Ewing sarcoma.
Additional attention has focused on features of physical growth and development that might distinguish patients with Ewing sarcoma. Differences in birth weight between patients with Ewing sarcoma and controls have not been observed. Given its peak incidence during adolescence, some groups have investigated differences in the onset of puberty between patients with Ewing sarcoma and controls. One group reported that boys with Ewing sarcoma began shaving earlier than boys without Ewing sarcoma. Other differences related to development of secondary sex characteristics have not been observed. At the time of diagnosis, patients with Ewing sarcoma do not seem to be taller than their peers.
Several viruses have been suggested as playing a role in the pathogenesis of Ewing sarcoma. One group reported that the adenovirus E1A gene could specifically induce the chromosomal translocation characteristic of these tumors. Follow-up studies failed to corroborate this finding. Based on increased Epstein-Barr virus titers in patients with Ewing sarcoma, other groups evaluated Ewing tumors for Epstein-Barr virus infection and found no viral involvement. Both BK and SV40 viral sequences have been variably identified in cases of Ewing sarcoma, but the functional importance of these findings remains unknown.
Ewing sarcoma rarely develops as a second malignancy. Several case reports describe the development of Ewing sarcoma years after the successful treatment of children with a range of hematologic malignancies and solid cancers. More formal studies indicate that Ewing sarcoma as a second malignancy occurs only infrequently. In a population-based study, 2.1% of all Ewing sarcoma cases arose as second malignancies. In a large institutional review, only 1.3% of second malignancies following treatment of pediatric cancer were Ewing sarcoma or PNET. Several large series of patients with secondary bone sarcomas have reported only a small number of patients with Ewing sarcoma. These secondary Ewing tumors do not appear to be radiation-related. In addition, the wide range of primary malignancies described in these patients suggests that these tumors do not arise as part of a specific cancer predisposition syndrome.
Biologic Features
Chromosomal Rearrangements in Ewing Sarcoma
In the mid-1980s the presence of a recurrent reciprocal chromosomal translocation, t(11;22)(q24;q12), was reported in Ewing sarcoma tumors and cell lines. The same translocation was also identified in PNET, supporting the hypothesis that these two tumors represented the same disease exhibiting varying levels of differentiation along a neuronal pathway. Subsequent studies demonstrated that t(11;22)(q24;q12) was present in approximately 83% of Ewing sarcomas.
The early analysis of translocations in Ewing sarcoma demonstrated that the derivative chromosome 22 was always maintained as these tumors and cell lines undergo clonal evolution, whereas the derivative chromosome 11 could be lost. This indicated that the “22;11” derivative is the key chromosomal abnormality, whereas the reciprocal “11;22” is unnecessary. The t(11;22)(q24;q12) is a tumor-specific or somatic mutation because it is not found in the germline DNA of patients.
In addition to the t(11;22)(q24;q12) rearrangement (and related translocations with similar molecular consequences, discussed later), other chromosomal abnormalities have also been reported in Ewing sarcoma. A careful analysis of these sometimes complex karyotypes has revealed a small group of recurrent, nonrandom chromosomal abnormalities. Gains (usually trisomies) of chromosome 8 may be observed in up to 50% of cases, and gains of chromosomes 12 and 1q are seen in approximately 25% of cases. Gains in chromosome 20 have also been reported in 10% to 20% of cases. Interestingly, a chromosomal translocation that is unrelated to the t(11;22)(q24;q12) has been observed in approximately 20% of cases: der(16)t(1;16). This translocation is often found as an unbalanced rearrangement and thus results in partial gains (trisomies and occasionally tetrasomies) of 1q and losses (monosomies) of 16q. The breakpoints of this translocation appear to be variable and range from q11 through q32 on chromosome 1 and from q11.1 through q24 on chromosome 16. This suggests that the translocation does not alter particular target genes at the breakpoint but rather introduces gains of 1q and losses of 16q. Finally, losses at 1p36 have also been observed. More recent high-resolution copy number studies have identified copy number alterations in specific genes or regions of the genome, although the role of specific genomic gains or losses has yet to be determined. There has also been a report of genomic instability, as measured by microsatellite instability and loss of heterozygosity, in Ewing sarcoma. This finding, however, is somewhat controversial, insofar as a second group was not able to replicate the microsatellite instability findings.
The Cloning of EWS/FLI
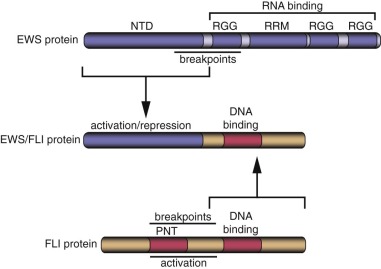
A major advance in the understanding of the pathogenesis of Ewing sarcoma occurred when the breakpoint of the (11;22)(q24;q12) translocation was cloned in 1992. The translocation breakpoint was localized roughly in the middle of two genes: EWSR1 and FLI1 . EWSR1 had not been previously identified and so was named on the basis of its involvement in the Ewing sarcoma translocation breakpoint ( Ew ing s arcoma r earrangement domain 1 ). FLI1 had been previously identified in mice as the F riend L eukemia Virus i ntegration site. As a result of the translocation event, these two genes become fused and the expressed transcript encodes the EWS/FLI fusion protein ( Fig. 61-3 ).
Wild-Type EWS Protein
EWSR1 encodes the EWS protein (see Fig. 61-3 ). Wild-type EWS contains 656 amino acids and appears to be ubiquitously expressed. EWS is primarily localized to the nucleus, although it has also been detected in the cytoplasm and on the cell surface. The amino terminus of EWS (sometimes referred to as NTD-EWS) contains 285 amino acids consisting of 31 pseudorepeats rich in tyrosine, glutamine, serine, threonine, glycine, alanine, and proline. This region bears some similarity to the carboxyl-terminal domain (CTD) of RNA polymerase II. This similarity suggested that the region could modulate RNA transcription. The carboxyl terminus of EWS contains an RNA recognition motif (an RRM domain), as well as three arginine-glycine-glycine (RGG) domains that are often found in RNA binding proteins. Indeed, it has been demonstrated, in vitro, that EWS can bind to RNA. These findings suggest that wild-type EWS may be involved in RNA transcription or processing. The carboxyl-terminal region also contains a potential C 2 C 2 zinc finger of uncertain significance. There is evidence that EWS may undergo posttranslational modifications, but the significance of these alterations is not well understood.
EWS is closely related to two additional sarcoma-associated translocation partners, TLS ( t ranslocated in l ipo s arcoma; also called FUS) and TAF15 (also known at TAF II 68, TAF2N, and RBP56), as well as to a Drosophila melanogaster protein named cabeza (also known as SARFH; originally called P19). TLS was first identified in human myxoid liposarcoma in the context of the t(12;16)(q13;p11) translocation, which encodes a TLS/CHOP fusion protein ( Table 61-1 ). TAF15 is a member of the TFIID general transcription complex. It is found fused to NR4A3 (also called CHN, TEC, CSMF, or NOR1) in extraskeletal myxoid chondrosarcomas and to ZNF384 (also called CIZ or NMP4) in acute leukemias (see Table 61-1 ). TAF15 also has significant homology and domain organization to EWS and TLS. The three human proteins are sometimes described as the TET family of proteins ( T LS, E WS, T AF15) or the FET family ( F US, E WS, T AF15). Although the normal function of these proteins is not completely understood, some evidence suggests that they may be involved in RNA splicing, gene expression, microRNA processing, and formation of RNA-containing intracellular granules. Interestingly, constitutional mutations in TLS result in familial amyotrophic lateral sclerosis (ALS), and mutations in TAF15 may also be associated with the disease.
| Tumor Type | Translocation | Fusion Gene |
|---|---|---|
| Ewing sarcoma | t(11;22)(q24;q12) | EWSR1/FLI1 |
| t(21;22)(q22;q12) | EWSR1/ERG | |
| t(7;22)(p22;q12) | EWSR1/ETV1 | |
| t(17;22)(q12;q12) | EWSR1/ETV4 | |
| t(2;22)(q35;q12) | EWSR1/FEV | |
| t(16;21)(p11;q22) | TLS/ERG | |
| t(2;16)(q35;p11) | TLS/FEV | |
| Ewing-like sarcoma | t(20;22)(q13;q12) | EWSR1/NFATC2 |
| (NB: can occur in ring chromosome and may be amplified) | EWSR1/POU5F1 | |
| t(6;22)(p21;q12) | EWSR1/SMARCA5 | |
| t(4;22)(q31;q12) | EWSR1/ZSG | |
| Submicroscopic inv(22) in t(1;22)(p36.1;q12) | ||
| t(2;22)(q31;q12) | EWSR1/SP3 | |
| t(4;19)(q35;q13) | CIC/DUX4 | |
| inv(X)(p11.4;p11.22) | BCOR/CCNB3 | |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWSR1/ATF1 |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1/WT1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWSR1/NR4A3 |
| t(9;17)(q22;q11) | TAF15/NR4A3 | |
| t(9;15)(q22;q21) | TCF12/NR4A3 | |
| Myxoid liposarcoma | t(12;16)(q13;p11) | TLS/DDIT3 |
| t(12;22)(q13;q12) | EWSR1/DDIT3 | |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11) | TLS/ATF1 |
| Low grade fibromyxoid sarcoma | t(7;16)(q33;p11) | TLS/CREB3L2 |
| Acute myelogenous leukemia | t(16;21)(p11;q22) | TLS/ERG |
| Acute myelogenous, lymphoblastic, or undifferentiated leukemia | t(12;22)(p13;q12) | EWSR1/ZNF384 |
| t(12;17)(p13;q11) | TAF15/ZNF384 |
* Gene names are provided rather than their corresponding protein names.
Wild-Type FLI Protein
FLI is a member of the ETS family of transcription factors. This family is defined by the presence of an ETS type DNA-binding domain and consists of 27 different members in humans. As with most transcription factors, ETS family members have a modular architecture with separate domains that contribute DNA binding and transcriptional functions. The DNA-binding ETS domain of FLI is present in the carboxyl-terminal portion of the protein (see Fig. 61-3 ). Most ETS family members (including FLI) bind to sequences containing a GGAA or GGAT core sequence. In addition to DNA-binding function, FLI contains two separate transcriptional activation domains, one in the amino terminus and a second in the carboxyl terminus (distal to the ETS DNA-binding domain; see Fig. 61-3 ). Wild-type FLI also contains a PNT (pointed) domain in the amino terminal portion of the protein. PNT domains are thought to mediate protein-protein interactions.
The normal role of FLI appears to be primarily in the hematopoietic lineage, where it regulates megakaryocytic development. Knockout of murine Fli-1 results in abnormal megakaryocytic differentiation and a loss in vascular integrity that results in embryonic lethality from central nervous system hemorrhage. Hemizygous deletion of FLI is found in patients with Paris-Trousseau or Jacobsen thrombocytopenia syndromes. FLI also appears to be important for vascular development in the mouse and zebrafish. FLI is also expressed in neural crest–derived mesenchyme in both the mouse and quail. This finding is particularly interesting in light of the neural crest phenotype of Ewing sarcoma.
FLI itself can function as an oncogene, at least in the hematopoietic context. Indeed, the murine Fli locus was first defined as a predominant F riend mu rine l eukemia v irus (F-MuLV) insertion site. Thus insertion of F-MuLV at the Fli locus results in upregulation of the Fli gene and subsequent development of erythroleukemia. The oncogenic activity of FLI appears to be limited to the hematopoietic system as introduction of the protein into other models of oncogenesis, such as the NIH3T3 cell model, does not result in the transformed phenotype.
EWS/FLI and Other TET/ETS Fusions in Ewing Sarcoma
EWS/FLI consists of the amino terminus of EWS fused, in frame, to the carboxyl terminus of FLI (see Fig. 61-3 ). The EWS portion retained in the fusion contains the pseudorepeat domain that harbors a strong transcriptional activation function as well as a transcriptional repression function. The RNA-binding domain of EWS is lost in the fusion protein and instead is replaced by the portion of FLI that contains its ETS DNA-binding domain.
EWS/FLI functions as an oncoprotein. EWS/FLI can induce NIH3T3 cells (an immortalized mouse fibroblast cell line) to exhibit both anchorage independent growth and growth as tumors when injected subcutaneously into immunodeficient mice. Conversely, blockade of EWS/FLI expression or function (via antisense RNA, dominant-negative blocking alleles, or RNA interference) causes Ewing sarcoma cells to lose their transformed phenotype. These data demonstrate that EWS/FLI expression is required for the oncogenic phenotype of Ewing sarcoma. Interestingly, it has been reported that after therapy-induced neural differentiation of Ewing sarcoma, EWS/FLI expression may also be lost. Although these data support an important role for EWS/FLI expression in Ewing sarcoma proliferation and transformation, they do not address whether EWS/FLI formation is the first step in Ewing sarcoma development or whether other oncogenic events occur first and are followed by EWS/FLI formation.
There are three main consequences of the t(11;22)(q24;q12) rearrangement. First, it places the transcriptional regulation of the EWS/FLI fusion under the control of the EWSR1 promoter. This is important because the activity of the FLI1 promoter is limited to hematopoietic (and perhaps neural crest) lineages, whereas the EWSR1 promoter appears to be active in most, if not all, cell types (see previous sections). Second, the translocation results in a loss of one wild-type EWSR1 allele and one wild-type FLI1 allele. Loss of one FLI1 allele is probably of no consequence because wild-type FLI1 is not expressed in Ewing sarcoma. Whether loss of one EWSR1 allele is important in Ewing sarcoma development is not known. The third consequence of the translocation is that it creates the EWS/FLI fusion protein. This is important because although EWS/FLI functions as an oncoprotein, neither wild-type EWS nor wild-type FLI could induce transformation of NIH3T3 cells, thus demonstrating a gain of function for the EWS/FLI fusion. Structure-function analyses of EWS/FLI demonstrated that both the amino terminal EWS domain and the ETS DNA-binding domain of FLI were required for oncogenic transformation of NIH3T3 cells and Ewing sarcoma cells. These results indicate that the fusion protein functions as an aberrant transcription factor. Comparisons of EWS/FLI with wild-type FLI demonstrated that the EWS domain included in the fusion protein could function as a much stronger transcriptional activation domain than the domain of FLI that was lost in the fusion. These results suggested that EWS/FLI induced oncogenic transformation by binding to target genes through its ETS DNA-binding domain and upregulating the expression of those genes through its EWS domain.
The mechanisms by which the EWS portion of the fusion functions as a transcriptional activation domain are only now beginning to be understood. Because wild-type EWS has been shown to interact with TFIID, RNA polymerase II, or the p300/CBP coactivators, it appears likely that these interactions are also important for EWS/FLI function. RNA helicase A (RHA) has also been shown to be important for the transcriptional activity of EWS/FLI. Thus RHA may also function as a coactivator for gene expression mediated by the fusion protein.
More recently, it has been appreciated that EWS/FLI also functions as a transcriptional repressor. For example, analysis of the transcriptional program triggered by EWS/FLI in Ewing sarcoma cells has revealed that more genes are downregulated by the fusion than are upregulated. Additional analysis has demonstrated that EWS/FLI functions as a direct transcriptional repressor at some target genes, like LOX and TGFBR2 . The mechanism of repression appears to be through binding of a transcriptional corepressor complex called NuRD, that includes nucleosome remodeling function, as well as histone deacetylase and histone lysine-specific demethylase activities. Both transcriptional activation and repression functions are required for the oncogenic activity of EWS/FLI in Ewing sarcoma cells.
Analysis of EWS/FLI binding sites in the human genome revealed that the fusion protein binds to sites that contain “classic” ETS-type high-affinity sequences (e.g., ACCGGAAGTG). However, a more interesting and unusual finding was that EWS/FLI was also found to bind to GGAA-containing microsatellite sequences. EWS/FLI binds to these microsatellites and uses them as response elements to activate the expression of target genes, and the transcriptional function is proportional to the length of the microsatellite. This raises the intriguing possibility that polymorphisms in GGAA-microsatellites associated with key EWS/FLI target genes (e.g., NR0B1, CAV1, or GSTM4 ) may alter susceptibility to Ewing sarcoma or perhaps outcome in patients with the disease. It has also been suggested that the lack of GGAA-microsatellites near critical EWS/FLI target genes, such as NR0B1 , in organisms other than humans might be the reason that only humans develop Ewing sarcoma.
In addition to EWS/FLI, other less frequent Ewing sarcoma translocation breakpoints were cloned and revealed a very similar structural organization (see Table 61-1 ). As described, approximately 83% of Ewing sarcoma tumors contain an (11;22)(q24;q12) translocation. Approximately 10% of cases demonstrate an alternate translocation, t(21;22)(q22;q12). Cloning of the breakpoint of this rearrangement revealed that EWS was fused, in frame, to another member of the ETS family, ERG. As in the case of EWS/FLI, the EWS/ERG fusion contains the amino terminus of EWS fused to the carboxyl terminus of ERG, which harbors its ETS DNA-binding domain. Interestingly, the ETS DNA-binding domains of FLI and ERG are 98% identical at the amino acid level. Subsequent investigations demonstrated that EWS/ERG also functioned as an oncoprotein in the NIH3T3 cell model. In subsequent years additional translocations were identified in Ewing sarcoma, including a t(7;22)(p22;q12), t(17;22)(q12;q12), and t(2;22)(q33;q12). Cloning of transcripts from these translocations demonstrated that in each case EWS was fused to another member of the ETS family. Currently, five EWS/ETS fusions have been described (see Table 61-1 ). It is assumed that all EWS/ETS fusion proteins bind similar (if not identical) target genes to induce oncogenic transformation.
Overall, most cases of Ewing sarcoma appear to harbor fusions between EWS and various ETS family members. Rarely, fusions between TLS and ETS family members may be present in Ewing sarcoma instead, as shown by the identification of TLS/ERG fusions in four patients with Ewing sarcoma and the identification of a TLS/FEV fusion in another case (see Table 61-1 ). These findings highlight the similarities across the TET family and across the ETS family. Perhaps the most generic manner of describing the fusion proteins in Ewing sarcoma would be as “TET/ETS fusions.” Because EWS/FLI is the most common fusion identified in Ewing sarcoma, most of the experimental work has been performed using this fusion protein. Experimental findings based on EWS/FLI have only occasionally been confirmed using other Ewing sarcoma fusion proteins.
In addition to variability of fusion partners, variability also exists in the exonic structure of some of the fusion proteins ( Fig. 61-4 ). The breakpoints found in Ewing sarcoma translocations occur in the introns of EWSR1 and FLI1 (and presumably the other ETS factors as well, although in most cases this has not been experimentally confirmed). The breakpoints in the EWSR1 gene are found in one of two regions (3 kb and 1.2 kb in size) present in an approximately 6 kilobase (kb) region that includes four of the gene’s 16 introns. The breakpoints in FLI1 occur over an approximately 50-kb region, which includes six of eight FLI introns. Splicing events join adjacent exons together in the fusion transcript. As a result of this variability in breakpoint and subsequent RNA splicing, at least 10 different EWS/FLI isoforms are generated (see Fig. 61-4 ).
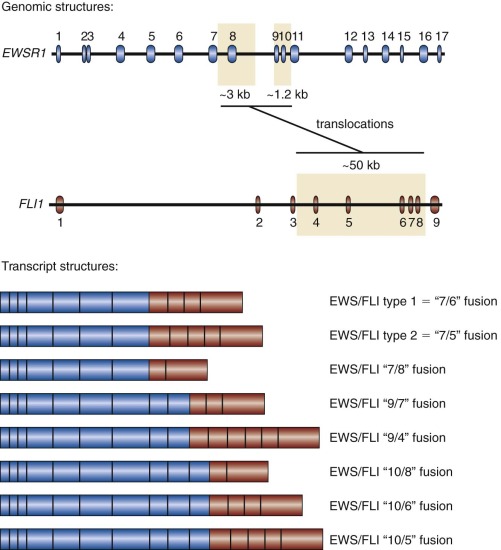
The most common Ewing sarcoma translocation fuses exon 7 of EWSR1 to exon 6 of FLI1 . This “7/6” fusion is commonly referred to as a “type 1” EWS/FLI fusion and accounts for approximately 50% to 60% of EWS/FLI fusions. A “type 2” fusion joins exon 7 of EWSR1 to exon 5 of FLI1 and is the second most common EWS/FLI fusion, accounting for approximately 20% to 30% of EWS/FLI fusions. Laboratory studies suggested that the type 1 EWS/FLI fusion was a weaker oncoprotein than the type 2 fusion, and originally it was thought to have prognostic significance. However, these functional differences were not reflected on changes in gene expression between type 1 and type 2 fusions, and large-scale clinical correlative data did not indicate a prognostic difference based on fusion subtype.
Although there is general agreement regarding the designation of the type 1 and type 2 fusions, there is no generalized nomenclature for the remaining fusions, and so these are best described in terms of which exons are fused to one another. In addition to variability in the EWS/FLI breakpoint, EWS/ERG has also been reported to demonstrate variability based on fusion points, with at least four fusions reported. Because the other fusions are so rare, there is only minimal information as to whether they also show similar levels of breakpoint heterogeneity.
Ewing Sarcoma Cell of Origin
The cell of origin of Ewing sarcoma is unknown, and the subject has generated intense debate. The original description by James Ewing in 1921 suggested that this “round cell sarcoma” was of endothelial origin and hence properly referred to as a “diffuse endothelioma of bone.” Dr. Ewing later suggested that it may arise from perivascular lymphatic endothelium. A hematologic origin was proposed in the early 1970s following an in-depth evaluation of light and electron microscopic characteristics and cytochemistry of both primary tumor and cell culture specimens. In the early 1980s a fibroblastic-mesenchymal derivation was proposed based on, among other things, the patterns of collagen expression of the tumor. In the late 1980s, however, a neural crest derivation was proposed because of the occasional identification of certain neural features, such as the presence of Homer Wright rosettes, neural processes, neurosecretory granules, and neural immunohistochemical markers. Additionally, undifferentiated Ewing sarcoma cell lines can occasionally be induced to express markers of neural differentiation, including the expression of neuritelike elongated processes, neuron-specific enolase (NSE), cholinesterase, and neural filament triplet protein. Interestingly, there are a few examples of neural differentiation of Ewing sarcoma occurring after therapy, which further supports this theory. More recently, it was shown that introduction of EWS/FLI into murine mesenchymal stem cells could result in oncogenic transformation, suggesting that these may be the cell of origin (although the same does not hold true for human mesenchymal stem cells). This hypothesis was further supported by microarray and cell biological studies performed on Ewing sarcoma cell lines in which EWS/FLI expression was reduced using RNA interference techniques. In the absence of EWS/FLI expression, these cells had a gene expression pattern that was reminiscent of mesenchymal stem cells and a cellular structural organization and adhesive and migratory properties that were also consistent with a mesenchymal phenotype. Furthermore, Ewing sarcoma cells with reduced EWS/FLI expression could be induced to differentiate into other mesenchymal cell types, such as fat and bone. This is a characteristic that is shared by mesenchymal stem cells.
It is important to note, however, that the phenotype of Ewing sarcoma (at least the neural crest component) may be a consequence of the (11;22) translocation rather than being related to the tumor’s cell of origin. Introduction of EWS/FLI into NIH3T3 murine fibroblasts induces a phenotype that is highly similar to Ewing sarcoma, including the small round blue cell morphology and features of neural differentiation. Similarly, expression of EWS/FLI in human rhabdomyosarcoma cells, neuroblastoma cells, or even normal human fibroblasts causes those cells to express genes that are similar to the genes expressed in Ewing sarcoma. This suggests that EWS/FLI may cause transdifferentiation of its target cells. Thus the neural crest phenotype of Ewing sarcoma may be induced by EWS/FLI.
Transcriptional profiling experiments (e.g., those using oligonucleotide or complementary DNA microarrays and, more recently, RNA sequencing) in Ewing sarcoma tumors and cell lines have allowed for a comprehensive analysis of the gene expression patterns associated with this disease. One of the earliest studies compared Ewing sarcoma tumors and cell lines with other small round cell tumors of childhood, including neuroblastoma, non-Hodgkin lymphoma, and rhabdomyosarcoma. These studies demonstrated that these histologically similar tumors could be distinguished from one another using gene expression data. A more recent analysis compared the expression of approximately 180 sarcoma tumor samples and again showed that tumors could be distinguished using gene expression patterns. Additional studies in which the gene expression pattern of Ewing sarcoma was compared with that of a variety of normal tissues supported the neural crest derivation but also suggested some similarity to endothelial cells. As discussed in the preceding section, analysis of some model systems has suggested a mesenchymal stem cell origin. A definitive understanding of the cell of origin of these tumors will require additional studies. It should be noted that as of yet no genetically engineered mouse model of Ewing sarcoma has been reported, and therefore tissue-specific oncogene expression or lineage-tracing approaches have not been used to identify the Ewing sarcoma cell of origin.
EWS/FLI Target Genes
Because most studies have suggested that EWS/FLI functions as a transcription factor, the two biggest questions in the field have been (1) what genes are dysregulated by EWS/FLI and (2) what are the roles of these gene targets in oncogenic transformation? These questions are beginning to be answered. Because the cell of origin of Ewing sarcoma is unknown, most laboratory-based studies of EWS/FLI have used heterologous cell types with varying results. More recent approaches have benefited from the use of “knockdown” (reduction) of gene expression using RNA interference (RNAi). The RNAi technique has allowed for the analysis of EWS/FLI in patient-derived Ewing sarcoma cell lines themselves, thus avoiding the concerns associated with using heterologous cell types.
Gene expression analysis of Ewing sarcoma cells in which EWS/FLI has been knocked down has allowed a comprehensive identification of the thousands of genes that are modulated by the fusion protein. For example, increased expression of NKX2.2 and NR0B1 are required for the oncogenic phenotype of Ewing sarcoma, although the specific mechanisms by which these transcriptional regulators facilitate the transformed phenotype remains poorly understood. Similarly, repression of TGFBR2 (the receptor for transforming growth factor β) and lysyl oxidase (LOX) by EWS/FLI are also required for tumorigenicity of Ewing sarcoma cells.
Gene expression studies also demonstrated that EWS/FLI represses the expression of the insulin-like growth factor binding protein 3 (IGFBP-3). IGFBP-3 appears to be involved in Ewing sarcoma apoptosis, and thus decreased expression of this protein appears to prevent apoptosis. These data complement a growing body of literature that supports a critical role for IGF-1 signaling in Ewing sarcoma development. For example, it was shown that Ewing sarcoma tumors and cell lines express IGF-1 and IGF receptors. IGF-1 and its receptor appear to function in an autocrine/paracrine fashion in Ewing sarcoma, and disruption of this signaling pathway blocks various functions that contribute to tumorigenesis. This occurs both in Ewing sarcoma cell lines as well as in model systems. Indeed, disruption of this pathway has shown some efficacy as a molecularly targeted therapeutic approach for this disease (discussed later).
NPY1R was identified as an EWS/FLI target gene through microarray analysis. Neuropeptide Y (NPY) is a small polypeptide neurotransmitter with diverse functions. Engagement of NPY receptors by NPY in Ewing sarcoma cell lines is growth inhibitory, suggesting a potential therapeutic role for these receptors. This result was surprising, insofar as Ewing sarcoma cells also express the NPY ligand. It appears likely that Ewing sarcomas are protected from the growth-inhibitory effect of NPY/NPY1R interactions by the expression of dipeptidyl peptidase IV, which cleaves NPY into a form that is unable to bind NPY1R. This cleaved form, NPY 3-36 , functions as an angiogenic factor by binding to NPY2R receptors on endothelial cells, thus providing a likely explanation for the maintenance of this growth-inhibitory autocrine pathway in Ewing sarcoma cells.
Another protein that has been shown to be upregulated by EWS/FLI is vascular endothelial growth factor (VEGF). VEGF levels are increased in Ewing sarcoma cell lines and xenografts. VEGF levels are also increased in the blood of patients with Ewing sarcoma and VEGF has been shown to be expressed in 55% of Ewing sarcoma tumor samples by immunohistochemistry. In the case of Ewing sarcoma, VEGF may induce not only angiogenesis but also vasculogenesis. Blockade of VEGF function in Ewing sarcoma model systems prevents tumor growth. These preclinical studies demonstrate the potential for VEGF inhibition as a therapeutic strategy for Ewing sarcoma.
Most, but not all, studies of Ewing sarcoma have shown that these tumors typically express telomerase activity. Interestingly, it appears that hTERT, the catalytic subunit of the telomerase holoenzyme, is upregulated by EWS/ETS proteins. This upregulation appears to be caused by specific binding of the ETS portion of the fusion to the TERT promoter. These data suggest that EWS/ETS fusions regulate telomerase activity by direct activation of the TERT promoter.
One key protein whose expression may be regulated by EWS/FLI is CD99 (also called MIC2). In human fibroblasts engineered to express EWS/FLI, the expression of CD99 closely mimicked that of the fusion protein. In the early 1990s it was found that CD99 is expressed at high levels in Ewing sarcoma cells but is uncommonly found on other tumor types. The CD99 antigen is recognized by a variety of monoclonal antibodies, including 12E7, HBA71, and O13, and these antibodies have an important role in the diagnosis of Ewing sarcoma (discussed later). The CD99 antigen is a membrane glycoprotein that plays a role in T-cell development and activation and migration of monocytes through endothelial junctions. In Ewing sarcoma CD99 inhibits neural differentiation and thus contributes to oncogenic transformation. Significantly, emerging data suggest that antibody-mediated engagement of CD99 may eventually be exploited as a new therapeutic approach to Ewing sarcoma. CD99 engagement in Ewing sarcoma results in apoptosis of the tumor cells. This effect enhances tumor cell killing by chemotherapeutic agents. Gene expression studies of CD99-mediated apoptosis suggested a role for the cytoskeletal protein zyxin in this process, and this finding was confirmed through functional studies. The identification of zyxin in this process was interesting in light of subsequent studies demonstrating that zyxin acts as a tumor suppressor in Ewing sarcoma. Taken together, these studies suggest that CD99 may be an effective therapeutic target in Ewing sarcoma by modulating cytoskeletal actin function.
Cooperative Pathways in Ewing Sarcoma Oncogenesis
Multiple potentially cooperating mutations in addition to TET/ETS fusions have been identified in Ewing sarcoma. The most widely assessed are mutations in the p53 and RB pathways. The p53 pathway includes proteins such as p14 ARF and MDM2, as well as p53 itself. When expressed in some heterologous systems, such as primary human fibroblasts, EWS/FLI induces the expression of p53. This finding suggests that there is selective pressure to inhibit the p53 pathway. This hypothesis appears to be at least partially true. Deletions in the CDKN2A locus (commonly called the INK4a locus encoding both the p14 ARF and p16 INK4a proteins; note that p14 ARF is involved in the p53 pathway, whereas p16 INK4a is involved in the RB pathway) have been identified in approximately 10% to 30% of primary Ewing sarcoma tumors and more than 50% of Ewing sarcoma cell lines. Amplifications or overexpression of MDM2 is occasionally seen in Ewing sarcoma. Mutations in p53 are found in 5% to 20% of Ewing sarcomas. Taken together, mutations in the p53 pathway may be present in nearly 70% of Ewing sarcomas, and these mutations may have prognostic significance in this disease (discussed later). Because there are likely additional components of the pathway yet to be identified or analyzed, the frequency of alterations in this pathway may be even higher.
The RB pathway regulates the cell cycle and includes proteins such as CDK4, D-type cyclins, and p16 INK4a . Mutations have been found in RB itself, and deletions of the INK4a locus have been documented as well in Ewing sarcoma. As was observed in the case of the p53 pathway, mutations in the RB pathway also appear to be relatively common in this disease.
Whereas mutations in the p53 and RB pathways are relatively common in Ewing sarcoma, mutations in other genes or pathways have only rarely been identified. For example, mutations in BRAF (which is also a member of the RAS/MAPK pathway) have been identified in 1 of 22 cell lines examined. No mutations in other components of the pathway, including RAS or receptor tyrosine kinases, have been identified, as of yet. This suggests that the RAS/MAPK pathway is only rarely activated by mutation in this disease.
Although the RAS/MAPK pathway does not appear to be frequently activated by mutation, there is some evidence for a requirement of pathway activation in Ewing sarcoma tumor development. In the NIH3T3 model system, it was shown that expression of EWS/FLI induced phosphorylation of the ERK1 and ERK2 proteins (downstream members of the MAPK pathway). This effect may result from autocrine stimulation of these transformed cells by PDGF-C, which was also shown to be induced by EWS/FLI expression in the NIH3T3 model, and expressed in Ewing sarcoma cell lines and tumor specimens. There is some controversy as to whether PDGF-C plays a role in the human disease, as PDGFR-α, which is required for PDGF-C responsiveness, is not expressed in Ewing sarcoma. Although PDGF-C may not be involved in Ewing sarcoma, inhibition of the MAPK pathway does block oncogenic transformation mediated by EWS/FLI in the NIH3T3 model. This finding was not replicated in Ewing sarcoma cells, thus raising the question as to whether this is an NIH3T3-specific pathway.
Many members of the Wnt signaling pathway are expressed in Ewing sarcoma, including soluble Wnt ligands, Wnt receptors (frizzled), and Wnt coreceptors (LRP5/6). Additionally, microarray analysis of EWS/FLI target genes has demonstrated downregulation of the Wnt inhibitor DKK1 and upregulation of various Wnt signaling components. These data suggest that Wnt signaling occurs in Ewing sarcoma. In contrast, analysis of the pathway in Ewing sarcoma cell lines in tissue culture has not demonstrated evidence of active Wnt autocrine signaling. If the pathway is involved in Ewing sarcoma, it seems likely that it is important for processes such as migration and metastasis, rather than proliferation and transformation. Current studies have not addressed this possibility.
Finally, genes involved in chromosomal stability or chromatin remodeling have also been implicated in Ewing sarcoma. For example, the STAG2 protein helps regulate chromosomal separation during mitosis and was found to be deleted in a number of human tumor types, including Ewing sarcoma. It was suggested that this may be a mechanism of chromosomal instability in Ewing sarcoma and other tumors. Deletion of SMARCB1 (encoding the SMARCB1/INI1/SNF5 protein) was found in approximately 10% of Ewing sarcoma cases. SMARCB1 is a member of the SWI/SNF chromosome remodeling complex whose deletion is tightly associated with atypical teratoid/rhabdoid tumor. Whether SMARCB1-deleted Ewing sarcoma tumors represent a unique subset of Ewing sarcoma or instead indicate that alterations in chromatin remodeling complexes are widespread in Ewing sarcoma is currently unknown. Interestingly, a newly identified chromosomal rearrangement, t(4;22)(q31;q12), encodes an EWS/SMARCA5 fusion protein in tumors that appear consistent with Ewing sarcoma, supporting the idea that alterations in this chromatin remodeling complex may indeed be widespread in this disease. Another possibility is that the EWS/SMARCA5 fusions are found in tumors that are similar, but not identical, to Ewing sarcoma. Indeed, the most recent World Health Organization guidelines have named these (and other non-TET/ETS translocation-bearing tumors that have similar appearance to Ewing sarcoma; see Table 61-1 ) as Ewing-like sarcomas.
Apoptotic Pathways in Ewing Sarcoma
As described previously, mutations in the p53 pathway are relatively common in Ewing sarcoma. These mutations likely confer resistance to the growth effects of TET/ETS fusions in the disease and may also protect against apoptosis. Whereas p53 is an important mediator of apoptosis, other signaling pathways are involved as well. These other pathways have been evaluated in Ewing sarcoma in a number of studies.
Both Fas and the Fas ligand (FasL) are expressed in the majority of Ewing sarcomas. The status of the FasL in Ewing sarcoma is somewhat controversial; one study reported soluble FasL in Ewing sarcoma–conditioned media, whereas another study reported that the FasL is only found intracellularly and thus is not accessible to cell surface receptor. In terms of the receptor, Fas at the cell surface can be functional, but Ewing sarcoma cells can be divided into Fas-sensitive, Fas-inducible, and Fas-resistant groups. Fas-sensitive lines are killed by coincubation with a FasL-expressing effector cell. Fas-inducible lines are killed by this effector cell line only after pretreatment with interferon-γ (IFN-γ) or cycloheximide (or both). The Fas-resistant lines were resistant regardless of pretreatment. The differences between sensitive and resistant cells are not completely known but may involve differences in Fas expression or expression of proapoptotic or antiapoptotic mediators, such as BAD and BAR. Strategies to improve Fas-mediated killing of Ewing sarcoma cells have been investigated, such as allowing for accumulation of more FasL through the use of metalloproteinase inhibitors and upregulation of Fas by administration of interleukin-12.
In addition to Fas/FasL, evaluations of tumor necrosis factor–related apoptosis-inducing ligand and tumor necrosis factor α as inducers of Ewing sarcoma apoptosis have also been conducted, in vitro and in vivo, alone and in combination with other agents. Taken together, these data support a potential use of proapoptotic ligands in the treatment of Ewing sarcoma. However, the variability of the effects seen demonstrate the need for an ongoing mechanistic evaluation of these pathways in this disease.
Biological Features Summary
The identification of t(11;22)(q24;q12) and subsequent cloning of the EWS/FLI fusion protein represented a major advance in the understanding of the pathogenesis of Ewing sarcoma. EWS/FLI appears to function primarily as an aberrant transcription factor to dysregulate gene targets involved in the development of this disease. Multiple gene targets and cooperating molecular pathways have been identified. These represent a wide array of functions, including growth factor signaling, survival and apoptosis pathways, angiogenesis, cellular immortalization, and cellular differentiation. Despite these myriad molecular functions, a unified mechanistic understanding of Ewing sarcoma development has not yet been reached. Similarly, the cell of origin of the disease has not yet been clearly identified. Nonetheless, there is great hope that a detailed understanding of the molecular mechanisms involved in Ewing sarcoma development will lead to new diagnostic, prognostic, and therapeutic approaches for this disease.
Clinical Presentation and Diagnosis
Clinical Presentation
The most common presenting symptoms of Ewing sarcoma are pain, a palpable mass, or both. In one series 89% of patients with Ewing sarcoma reported pain at the time of initial presentation. Pain does not necessarily occur at night; this pattern was reported in only 19% of patients in one series. Patients may report that their pain began at the same time as a minor musculoskeletal injury. This presentation, reported in approximately 25% of patients, may delay the diagnosis. Unlike patients with osteosarcoma, patients with Ewing sarcoma may report constitutional symptoms such as fever and weight loss. Approximately 15% to 20% of patients have fever at presentation. Other symptoms depend on the site of disease. For example, 40% to 94% of patients with paraspinal tumors have presenting symptoms of spinal cord compression. Patients with large pelvic tumors may complain of an alteration in voiding. Extensive bone marrow metastatic disease may cause symptoms related to anemia, thrombocytopenia, or neutropenia. Patients may have symptoms for many weeks or months before seeing a physician for evaluation, with most studies reporting an average time from symptom onset to diagnosis of 3 to 5 months.
Ewing sarcoma can arise from any bone but has a predilection for pelvic bones and long bones of the leg. Approximately 45% of Ewing sarcomas develop in the axial skeleton. More than half of these axial tumors arise from pelvic bones, and a quarter of axial tumors arise from a rib. Up to 30% of Ewing sarcomas arise from long bones in the leg, and up to 15% of tumors develop in long bones in the arm. Tumors of the hands, feet, and head develop only rarely, accounting for 10% or fewer of all tumors. The distribution of primary tumor site does not vary among racial groups. Soft tissue tumors may arise in any location, although axial sites predominate.
Laboratory studies in patients with newly diagnosed Ewing sarcoma may reveal nonspecific abnormalities. More than a third of patients have an elevated erythrocyte sedimentation rate (ESR) at the time of diagnosis. Similarly, serum lactate dehydrogenase (LDH) levels are elevated in approximately one third of patients. Laboratory manifestations of tumor lysis syndrome are uncommon with these tumors. Unlike osteosarcoma, alkaline phosphatase is not typically elevated in patients with Ewing sarcoma of the bone. Patients with advanced bone marrow metastatic disease may have anemia, thrombocytopenia, and neutropenia detected on a complete blood count. In one large series 12% of patients had anemia even in the absence of metastatic disease.
Box 61-1 summarizes the differential diagnosis of Ewing sarcoma. In most cases of Ewing sarcoma of the bone, the main alternative diagnosis is osteosarcoma. Table 61-2 displays features that may help differentiate Ewing sarcoma of the bone from osteosarcoma. Compared with osteosarcoma, Ewing sarcoma is more likely to occur in the axial skeleton. Those Ewing tumors that arise in long bones have a greater tendency to occur in the diaphysis, compared with the tendency of osteosarcoma to arise in the metaphysis of long bones. Patients with Ewing sarcoma are more likely to report constitutional symptoms than are patients with osteosarcoma. Other malignant tumors in the differential diagnosis of Ewing sarcoma of the bone include primary bone lymphoma, giant cell tumor of the bone, and bone metastasis of an extraosseous malignancy. The differential diagnosis of Ewing sarcoma arising from the soft tissues is more broad because the site of origin for these tumors is not restricted to the bone. The site of origin may help narrow the differential diagnosis. Paraspinal tumors may be confused with neuroblastoma, particularly in younger patients. Pelvic tumors may suggest rhabdomyosarcoma or malignant germ cell tumor. Lymphoma and other soft tissue sarcomas can arise at any site and should be included in the differential diagnosis.
Ewing Sarcoma of Bone
Osteosarcoma
Primary bone lymphoma
Ewing-like sarcoma
Langerhans cell histiocytosis
Osteomyelitis
Metastasis of an extraosseous malignancy
Benign bone tumor
Osteoblastoma
Chondrosarcoma
Giant cell tumor of the bone
Ewing Sarcoma of Soft Tissue
Rhabdomyosarcoma
Nonrhabdomyosarcoma soft tissue sarcoma
Lymphoma
Benign soft tissue tumor
Ewing-like sarcoma
Neuroblastoma
Malignant germ cell tumor
| Ewing Sarcoma | Osteosarcoma | |
|---|---|---|
| Age distribution | Peak in adolescence Occurs in young children Very rare in adults >40 years | Peak in adolescence Very rare in children <5 years Occurs in older adults |
| Racial distribution | Rare in patients of African or East Asian ancestry | No racial predilection |
| Predisposing factors | Not associated with radiation No known familial predisposition | Radiation exposure Li-Fraumeni syndrome History of retinoblastoma |
| Constitutional symptoms | Yes | No |
| Involved bones | Both flat and long bones | Long bones more common |
| Location in long bones | Diaphyseal more common | Epiphyseal/metaphyseal more common |
| Type of periosteal reaction | Laminated/layered “Onion-skinning” | Spiculated “sunburst” |
| Laboratory findings | Normal alkaline phosphatase Abnormal CBC if marrow disease | Elevated alkaline phosphatase Normal CBC |
| Histologic findings | Small round blue cells No malignant osteoid | Malignant spindle cells Malignant osteoid |
Reports from several cooperative group studies have defined the metastatic behavior of Ewing sarcoma. Approximately 25% of patients exhibit distant metastases at presentation. Rates of metastatic disease do not appear to differ between patients with skeletal tumors and those with soft tissue primary tumors. Patients with pelvic primary tumors have an increased incidence of metastatic disease at initial diagnosis. The lung is the most common metastatic site, with pulmonary dissemination reported in 50% to 60% of patients with metastatic disease. Patients with isolated pulmonary metastasis represent between 26% and 36% of cases of metastatic disease. Patients usually have multiple lung nodules identified in both lungs.
The bone is the next most common metastatic site at presentation. One group reported cortical bone metastases in 43% of patients with metastatic disease. A large cooperative group reported that the bone marrow is involved in approximately 19% of metastatic patients at initial diagnosis. A smaller series noted bone marrow involvement in 52% of patients with metastatic disease. This group performed up to 10 bone marrow aspirates on patients at initial presentation, suggesting that routine sampling may underestimate the incidence of bone marrow involvement. Brain metastasis at initial diagnosis appears to be extremely uncommon. In two series of patients with brain metastasis from Ewing sarcoma, all cases were reported at the time of disease recurrence and not at initial presentation.
Fewer than 10% of patients have lymph node involvement at initial diagnosis. The incidence of node involvement is higher in patients with soft tissue rather than bone primary tumors. In one population-based study, the incidence of regional node involvement among patients with soft tissue tumors was 12.4% compared with 3.2% in patients with bone primary tumors. The incidence of regional node involvement is also higher among patients with distant metastatic disease.
This metastatic pattern helps determine the appropriate staging evaluation for patients with Ewing sarcoma ( Box 61-2 ). In addition to computed tomography (CT) or magnetic resonance imaging (MRI) scans of the primary tumor, all newly diagnosed patients should be evaluated with a chest CT scan. Assessment for bone metastasis has conventionally been with radiolabeled-technetium bone scan. More recent data indicate that positron emission tomography (PET) scans are superior for this indication (discussed later), prompting wider adoption of PET imaging at initial diagnosis. At least bilateral bone marrow aspirates and biopsies should routinely be performed in newly diagnosed patients, with biopsy more sensitive for detection of marrow involvement compared with aspirate. Attention to regional nodes on imaging studies is particularly warranted in patients with soft tissue primary tumors. These evaluations will indicate whether a patient has metastatic or nonmetastatic Ewing sarcoma at initial diagnosis. This distinction serves as the main staging classification in practice for these patients. Although a staging classification for musculoskeletal tumors has been devised by Enneking, this system is not routinely used in the clinical care of patients with Ewing sarcoma.
Assessment of Primary Tumor
Magnetic resonance imaging (MRI) scan and/or
Computed tomography (CT) scan
Evaluation for Metastatic Disease
CT scan of the chest
Whole body fluorodeoxyglucose positron emission tomography scan or radiolabeled-technecium bone scan
Bone marrow aspirate and biopsy
Evaluation Before Initiating Chemotherapy
Echocardiogram
Serum creatinine (with formal creatinine clearance if renal function in question)
Imaging Features
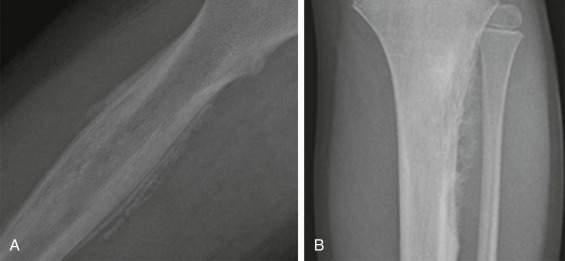
Most patients with Ewing sarcoma of the bone are initially evaluated with a plain radiograph. Two representative radiographs are shown in Figure 61-5 . These tumors typically appear as poorly circumscribed lesions arising from the bone, but with an associated soft tissue mass. Both lytic and sclerotic areas may be seen within the bony component of the tumor. Given the aggressive nature of these tumors, periosteal reaction is commonly observed. A layered or laminated periosteal reaction is most typical, often giving an onion-skin appearance (see Fig. 61-5, A ). A spiculated “sunburst” periosteal reaction is more commonly associated with osteosarcoma but may be occasionally observed in Ewing sarcoma (see Fig. 61-5, B ). Codman triangle, a triangular area forming under the periosteum in the setting of a rapidly growing bone lesion, may also be seen in some cases. Cortical thickening is present in approximately 20% of tumors. Pathologic fractures occur in 15% of patients with Ewing sarcoma of bone.
CT scans and MRI scans are used to better define the extent of local tumor in patients with both osseous and extraosseous tumors. CT scans most commonly demonstrate a heterogeneous mass with heterogeneous contrast enhancement. The soft tissue component generally has lower attenuation than muscle on non-enhanced CT scans. MRI scans typically reveal a mass that is heterogeneous with respect to both signal intensity and gadolinium contrast enhancement. Typically T1- and T2-weighted MRI scans demonstrate areas of increased signal intensity of the soft tissue component compared with skeletal muscle. Although both CT and MRI are able to delineate the soft tissue component of the tumor, MRI may be better able to determine the extent of bone marrow extension and the degree of growth plate involvement.
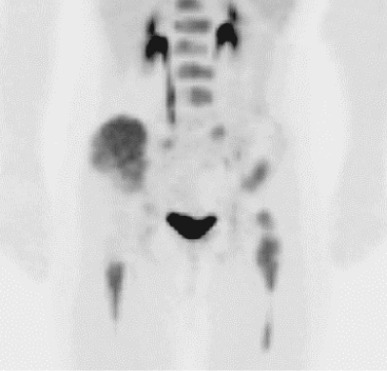
Emerging evidence suggests that fluorodeoxyglucose PET (FDG-PET) may play an increasing role in the management of patients with Ewing sarcoma. In one series all 32 patients with Ewing sarcoma evaluated by FDG-PET imaging before initiation of chemotherapy had FDG-avid tumors. Figure 61-6 demonstrates an FDG-PET scan of a patient with widely metastatic Ewing sarcoma at initial presentation. A group of studies has evaluated the role of FDG-PET imaging in screening for metastatic disease and for disease recurrence. Several groups have demonstrated that FDG-PET imaging has superior sensitivity and specificity compared with conventional bone scans for detecting bone metastases in patients with Ewing sarcoma. In contrast, FDG-PET appears to be inferior to spiral CT scans in screening for pulmonary metastases in these patients. In screening patients with Ewing sarcoma for overall disease recurrence at any site, FDG-PET imaging may have lower sensitivity but higher specificity compared with conventional imaging. Finally, one group has reported that a decrease in FDG-avidity in response to neoadjuvant chemotherapy correlates with improved progression-free survival.
Pathologic Diagnosis
Once imaging studies have localized the tumor, diagnostic tissue must be obtained. The current pathologic diagnosis of Ewing sarcoma depends on tumor morphology, immunohistochemistry findings, and demonstration of a Ewing sarcoma–specific translocation (by standard cytogenetics, fluorescence in situ hybridization [FISH], or reverse transcriptase polymerase chain reaction assays [RT-PCR]). In planning for biopsy, the physician must ensure that adequate tissue will be available to perform each of these tests. For example, one series reported successful cytogenetic analysis in only 59% of fine needle aspirations performed for Ewing sarcoma. The biopsy should be planned with an eye toward ultimate local control of the tumor. If surgical resection is ultimately planned, the biopsy tract should be included in the resected specimen.
Ewing sarcoma and PNET form a morphologic continuum ranging from tumors with no apparent neural differentiation to tumors with evidence of early neural differentiation. Of note, the histologic classification of these tumors does not imply site of origin because tumors with either histology may arise in the bone or soft tissue. Ewing sarcoma appears as relatively monomorphic small round blue-staining cells with scant cytoplasm ( Fig. 61-7 ). The nuclei tend to be round without prominent nucleoli. These cells demonstrate positive periodic acid–Schiff staining owing to cytoplasmic glycogen. Electron microscopy may reveal these glycogen stores. The tumor cells grow in sheets without additional structure. A large cell or atypical Ewing sarcoma subset has been described with large irregular nuclei and more conspicuous nucleoli. As many as 30% of tumors with Ewing sarcoma histology arise in soft tissues rather than bone. PNETs typically display primitive Homer Wright pseudorosette formation. Electron microscopy may reveal additional features suggestive of neural differentiation, such as neurosecretory granules. Tumors without pseudorosette formation may also be classified as PNETs if they express two or more neural markers (discussed later). Approximately 50% of tumors with PNET histology arise in bone. Despite these morphologic differences, Ewing sarcoma and PNET are now viewed as the same biological entity.
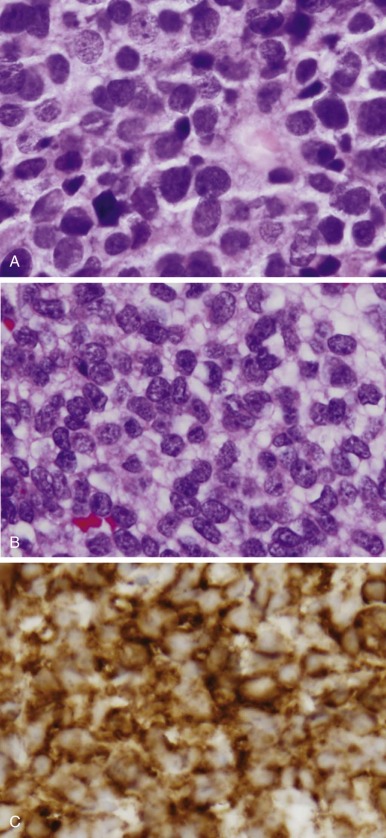
Immunohistochemistry plays a key role in differentiating Ewing sarcoma and PNET from other small round cell tumors of children and young adults. The most useful antigen in the diagnosis of Ewing sarcoma appears to be CD99. This protein product of the MIC2 gene is expressed in a limited number of normal tissues, including strong staining of ependymal cells, pancreatic islet cells, anterior pituitary gland, testicular Sertoli cells, ovarian granulosa cells, and maturing T lymphocytes. Less intense staining has been noted in scattered endothelial cells. Ewing sarcoma and PNET both display prominent CD99 immunostaining in a membranous pattern (see Fig. 61-7 ). Multiple series have found strong CD99 immunostaining in more than 90% of cases of Ewing sarcoma and PNET. Staining for neural markers, such as synaptophysin, S-100, NSE, and neurofilament, is variable and helps to characterize tumors as Ewing sarcoma or PNET. These tumors also commonly stain with vimentin, whereas desmin staining is distinctly uncommon. Approximately 30% of Ewing sarcomas and PNETs demonstrate diffuse c-kit (CD117) staining.
Several tumors also express CD99 and may confuse the diagnosis. Strong CD99 immunostaining has been observed in ependymoma, glioblastoma, and pancreatic islet cell tumors. These tumors are not typically within the clinical differential diagnosis of Ewing sarcoma or PNET. In contrast, lymphoblastic lymphoma (particularly T-cell), neuroblastoma, alveolar rhabdomyosarcoma, synovial sarcoma, and desmoplastic small round cell tumor may demonstrate variable levels of CD99 staining and are frequently considered in the differential diagnosis of these tumors. Whereas both Ewing sarcoma and lymphoblastic lymphoma may demonstrate strong CD99 staining, Ewing sarcoma and PNET should be negative for lymphocyte markers, particularly leukocyte common antigen and terminal deoxynucleotidyl transferase. Ewing sarcoma and PNET are also more likely to demonstrate strong vimentin staining compared with lymphoblastic lymphoma. The immunohistochemical differentiation of Ewing sarcoma and PNET from neuroblastoma is typically more straight-forward largely because CD99 staining in neuroblastoma, if present, is weak and patchy. Neuroblastomas commonly demonstrate strong staining with NSE, whereas Ewing sarcoma and PNET occasionally will show weak to moderate NSE staining. Poorly differentiated synovial sarcoma and desmoplastic small round cell tumor may both also demonstrate CD99 staining and morphologically be difficult to differentiate from Ewing sarcoma and PNET. In both cases strong membranous staining is more consistent with Ewing sarcoma and PNET. In contrast, weak staining for CD99 and strong staining for cytokeratin and epithelial membrane antigen suggest synovial sarcoma. Desmoplastic small round cell tumors reliably demonstrate nuclear staining for WT1, whereas Ewing sarcoma and PNET typically do not stain with WT1. Nuclear staining for FLI1 may also help differentiate these tumors from other pediatric CD99 small round cell tumors. Most cases of Ewing sarcoma and PNET demonstrate nuclear FLI1 staining, whereas synovial sarcoma, neuroblastoma, and rhabdomyosarcoma only rarely show positive nuclear FLI1 staining. Significantly, however, a high proportion of lymphoblastic lymphomas also exhibit nuclear FLI1 staining. Finally, a number of small round cell tumors of bone harboring novel translocations have been described (see Table 61-1 ). These have recently been classified as Ewinglike sarcomas. As an example, a BCOR/CCNB3- containing tumor has recently been described. This entity has a gene expression profile that is distinct from that of Ewing sarcoma, demonstrates membranous CD99 positivity in only a minority of cases, and can be identified by intense immunohistochemical staining for CCNB3.
The combination of characteristic morphology and immunohistochemistry typically allows an experienced pathologist to render the diagnosis of Ewing sarcoma or PNET. The identification of recurrent chromosomal translocations and their associated oncoproteins in these tumors has allowed for the incorporation of molecular testing into their diagnosis. These methods play a role as confirmatory tests in the setting of typical morphology and immunohistochemistry. In more unusual cases these molecular tests may serve as the key feature on which the diagnosis is based. Although these molecular approaches have been very effective, it is worth recognizing their strengths and weaknesses when interpreting diagnostic studies.
Karyotypic analysis is one approach to confirming the diagnosis of Ewing sarcoma. The presence of one of the characteristic translocations can be considered strong supportive evidence for the diagnosis. It is important to recognize that false-negative results may occur from a number of sources, including overgrowth of nonmalignant elements during culture, complex translocations in which the characteristic Ewing-associated rearrangement is partially masked by the presence of an additional fusion partner, and cryptic rearrangements in which the translocation cannot be identified using typical techniques. FISH can be very helpful as a complementary technique to demonstrate that the EWSR1 locus is “split,” which suggests a translocation event. FISH for the FLI1 and/or ERG loci that demonstrates a split signal from either of these genes or fusion between EWSR1 and FLI1 or ERG signals provides additional strong evidence for involvement of these genes in the rearrangement. A false-negative FISH result may occur when the fusion involves a rarer translocation partner, such as TLS (in lieu of EWS) or ETV1, ETV4, or FEV (in lieu of FLI or ERG). These rare translocations are not typically included in the molecular diagnostic evaluation of these tumors.
A positive EWSR1 “split” FISH assay is not specific for the diagnosis of Ewing sarcoma because of the presence of EWSR1 rearrangements in tumor types other than Ewing sarcoma (see Table 61-1 ). For example, clear cell sarcoma (also called malignant melanoma of the soft parts) usually harbors a t(12;22)(q13;q12) rearrangement. This translocation fuses EWS to the ATF1 protein to form the EWS/ATF1 protein. Similarly, desmoplastic small round cell tumor harbors a t(11;22)(p13;q12) rearrangement. This fuses the EWS protein to the Wilms tumor suppressor WT1 to form the EWS/WT1 protein. EWS or TAF15 can join the NR4A3 orphan nuclear receptor to form EWS/NR4A3 and TAF15/NR4A3 fusion proteins in extraskeletal myxoid chondrosarcoma. EWS is occasionally fused to DDIT3 (also called CHOP) to form EWS/DDIT3 in t(12;22)(q13;q12) positive myxoid liposarcoma. An EWS/ZNF278 fusion was identified in a small round cell sarcoma as a result of a t(1;22)(p36;q12) rearrangement. In some cases there may be diagnostic confusion between some of these tumor types and Ewing sarcoma, and thus the presence of a rearranged EWSR1 locus may not be adequate for diagnostic purposes. In rare instances tumors may show biphenotypic differentiation with both neural and myogenic patterns. At least some of these express EWS rearrangements (EWS/FLI or EWS/ERG fusions) as well as the classic PAX3/FKHR fusion protein associated with alveolar rhabdomyosarcoma.
The use of RT-PCR may provide greater specificity in the diagnosis of Ewing sarcoma. RT-PCR is a useful method for detection of EWS/ETS fusions. As with any PCR-based method, false-positive results may occur from contamination. False-negative results may occur because of degradation of the RNA, alternate fusion partners, or because an RT-PCR primer pair may be designed to detect some EWS/FLI transcripts but not others. False-negative results may also occur when biopsies do not capture viable tissue or primarily include nonmalignant elements. Despite their shortcomings, molecular techniques have become an important addition to the diagnostic approach for Ewing sarcoma.
Prognostic Factors
A number of clinical and molecular features have been identified as prognostic variables in patients with Ewing sarcoma. The presence of metastatic disease at initial diagnosis is widely regarded as the most important adverse clinical prognostic feature. In the largest analysis of prognostic features in this disease, patients with metastatic disease at presentation had a 5-year relapse-free survival rate of 21% compared with 55% for nonmetastatic patients. Patients in this analysis received therapy according to several sequential treatment protocols. Patients with metastatic disease treated in the North American study INT-0091 had a similar 5-year event-free survival rate of 22% regardless of their chemotherapy regimen. Patients with isolated lung metastases have a better outcome than patients with dissemination to other sites or with dissemination to lungs plus other sites. Within this group of patients, patients with unilateral lung metastases may fare better than patients with bilateral lung metastases. Dissemination to regional lymph node is also an adverse prognostic factor, even among patients with otherwise localized disease.
Tumor size has long been recognized as a predictor of outcome in patients with Ewing sarcoma. Patients with larger tumors are at increased risk for having metastatic disease at initial diagnosis. For example, a pooled analysis of German patients treated in the European Intergroup Cooperative Ewing’s Sarcoma Study (EICESS) and Cooperative Ewing’s Sarcoma Study (CESS) indicated that tumor volume greater than 100 mL was an independent predictor of metastatic disease at initial diagnosis. The effect of tumor size on outcome does not depend entirely on this increased risk of metastatic disease, however. Tumor size appears to be prognostic even among patients with initially nonmetastatic tumors. The CESS-81 study demonstrated that tumor volume greater than 100 mL was a significant adverse prognostic factor for patients treated with either surgery or radiation. The follow-up CESS-86 study found that tumor volume greater than 200 mL was a major adverse prognostic feature for patients with localized Ewing sarcoma. The North American INT-0091 study reported that nonmetastatic patients with tumors greater than 8 cm in maximal dimension had an inferior outcome compared with patients with smaller tumors. The French EW88 study reported that tumor size, using either tumor volume or longest dimension, was prognostic only among patients who received radiotherapy alone as the mode of local control. Although some smaller studies have not observed this effect of tumor size on outcome, most investigators consider tumor size prognostic. Indeed, a large pooled analyses of prognostic factors in Ewing sarcoma identified large tumor volume as an independent predictor of poor outcome.
Tumor site is also widely considered a prognostic factor in Ewing sarcoma, with axial tumors carrying a worse prognosis. The Intergroup Ewing Sarcoma Study 1 (IESS-1) established pelvic site as an unfavorable prognostic feature. Several other cooperative group studies have confirmed this finding and indicated that patients with axial tumors generally have a worse outcome compared with patients with nonaxial tumors. In many of these studies, patients with pelvic primary tumors have been shown to have a particularly poor outcome compared with patients with other axial locations. However, the inferior outcomes in patients with pelvic tumors may be confounded by a higher incidence of both large tumor size and metastatic disease. For example, in the CESS-86 trial, axial tumor location was a significant adverse prognostic feature on univariate analysis. After controlling for tumor size and chemotherapy response, however, the prognostic impact of axial site became insignificant. Several groups evaluated only patients with nonmetastatic tumors and found that patients with pelvic tumors still had an inferior outcome compared with patients with nonpelvic tumors. Two other groups used multivariate methods to evaluate the prognostic impact of axial tumor location in nonmetastatic patients. Both groups reported that axial tumors have an inferior outcome compared with extremity tumors, even after controlling for potential confounders. Neither analysis controlled for tumor size, however. Therefore whether axial tumor location portends poor outcome independent of large tumor size remains unclear.
The prognostic impact of soft tissue rather than bone primary tumor origin remains unclear. In one population-based study, soft tissue origin was independently associated with improved outcomes compared with bone origin. In contrast, a cooperative group clinical trial that included both patients with soft tissue and bone tumors showed equivalent outcomes. As more trials include patients with soft tissue tumors, the prognostic impact of tumor origin will become clearer.
A number of groups have specifically evaluated histologic response to neoadjuvant chemotherapy as a predictor of outcome. Patients with a favorable chemotherapy response (typically 90% to 100% tumor necrosis depending on the grading scale used) at the time of surgery have been found to have a decreased risk of disease recurrence compared with patients who exhibited less necrosis. For patients with nonmetastatic tumors of the extremity, chemotherapy response is prognostic independent of age and tumor size. Despite these results, grading of chemotherapy response in Ewing sarcoma has not yet become routine practice, perhaps at least in part because this variable is not evaluable in those patients who receive definitive radiotherapy as their mode of local control. An alternative to histologic grading is radiographic grading of chemotherapy response. Two groups have reported that favorable radiographic response to chemotherapy is a predictor of favorable outcome. Evaluation of radiographic response may also not be possible in all patients, particularly in patients with tumors confined to bone without an associated soft tissue mass.
Age at initial diagnosis has been increasingly recognized as an important prognostic feature in Ewing sarcoma. Several studies have indicated that younger patients have an improved outcome compared with older patients. Older patients have an increased risk of presenting with large tumors, but interestingly they do not have a higher incidence of metastatic disease at initial diagnosis. Even controlling for these potential confounders, age has been identified as an independent predictor of outcome in a number of studies. Most studies have used a single cut-point in the 12- to 15-year age range. One study divided patients into three groups (younger than 10 years, 10 to 17 years, and 18 years or older) and found that nonmetastatic patients 18 years of age or older at initial diagnosis had a particularly poor outcome (44% 5-year event-free survival rate). A number of studies have focused on outcomes in older adolescents and adults with Ewing sarcoma. These studies have typically identified the same adverse prognostic features (metastatic disease, large size, axial location, and poor chemotherapy response) in older patients that have been identified in larger studies in patients at a wide range of ages. The mechanism underlying these differences in outcome with age remains unclear. Possible explanations include biological differences in tumor and host as well as health care delivery differences in compliance and dose intensity. For example, at least two reports have noted higher rates of soft tissue tumors in adults older than 40 years of age at initial diagnosis, which suggests biological differences in this disease when it arises in older patients. Another series noted lower treatment intensity and inferior outcomes in older patients, whereas another series reported similar treatment intensity and outcomes in older patients. These results highlight the importance of optimizing treatment intensity to maximize outcomes in adults with this disease.
Serum LDH at initial diagnosis also appears to be prognostic. Several early studies reported that patients with elevated serum LDH levels at diagnosis had an increased risk of disease recurrence. More recent studies have confirmed this finding and have found that the negative prognostic impact of an elevated LDH remains even after controlling for other possible confounding variables. A meta-analysis evaluating a range of tumor markers in Ewing sarcoma also concluded that elevated LDH at diagnosis is associated with inferior outcomes. Presumably, elevated LDH levels indicate greater tumor cell turnover and therefore more aggressive clinical behavior.
A variety of other clinical and demographic features at diagnosis have been variably reported to be prognostic in Ewing sarcoma. Two groups have reported that female sex is an independent predictor of favorable outcome. Other groups have not corroborated this finding. Other studies have demonstrated superior outcomes among the U.S. white, non-Hispanic population compared with members of other racial and ethnic groups. A greater degree of neural differentiation has been reported as an unfavorable marker by some groups but not by others. * Fever, anemia, leukocytosis, elevated erythrocyte sedimentation rate, and hypoalbuminemia at initial diagnosis have also been suggested as unfavorable prognostic features, but their prognostic value has not yet been validated.
* References .
The nature of the Ewing sarcoma–specific translocation does not appear to provide prognostic information. One group has demonstrated that patients with any type of EWS/FLI translocation have similar outcomes to patients with any type of EWS/ERG translocation. Previous studies suggested that patients with a type 1 EWS/FLI translocation had better outcomes compared with other types of EWS/FLI or EWS/ERG translocations. However, two large cooperative group trials both demonstrated that translocation subtype does not affect survival in the setting of modern therapy.Several studies have measured EWS/FLI fusion transcripts in the peripheral blood or bone marrow to predict outcome. These studies use RT-PCR techniques to identify occult metastatic disease, with 25% of clinically nonmetastatic patients noted to have detectable EWS/FLI fusion transcripts in blood or bone marrow in one early study. The results of studies correlating these findings to clinical outcome have been mixed. In one report patients with clinically localized disease but positive peripheral blood EWS/FLI RT-PCR at diagnosis had a higher risk of relapse compared with patients with negative peripheral blood EWS/FLI RT-PCR. A second study evaluating bone marrow EWS/FLI RT-PCR at diagnosis was not able to confirm this finding. Patients in this study had follow-up EWS/FLI RT-PCR performed on blood or bone marrow (or both). Patients with positive blood or bone marrow EWS/FLI RT-PCR at their last evaluation had a significantly increased risk of clinical disease recurrence. The utility of this methodology for predicting outcome in patients with Ewing sarcoma will require further prospective evaluation.
Chromosomal changes beyond the characteristic TET/ETS translocations may also provide prognostic information. Although it is the most common additional karyotypic abnormality in Ewing sarcoma, at least three groups have reported that trisomy 8 does not appear to influence outcome. A small series of 21 patients reported that all three patients with gain of chromosome 1q died as a result of disease. Another study of 88 patients with complete cytogenetic data did not observe an association between 1q gain and poor outcome, although only 12 patients had 1q gain. A larger study of 124 patients with Ewing sarcoma included 26 patients with gain of chromosome 1q and found that patients with gain of chromosome 1q have a worse outcome. One study reported that more global karyotypic anomalies may also be prognostic, including complex karyotypes (more than five chromosomal anomalies) or hyperdiploidy (more than 50 chromosomes).
Aberrations in p53 and p16 have also been correlated with outcome in patients with Ewing sarcoma. The 10% to 15% of patients with mutations in the TP53 gene (encoding the p53 protein) have consistently been shown to have a significantly worse outcome compared with patients who have wild-type TP53 , even after controlling for other prognostic factors. Additional work has suggested that mutations or deletions of the CDKN2A/INK4A gene may also confer a poor outcome in these patients. For example, aberrant TP53 or CDKN2A/INK4A gene status in one study was shown to be an independent negative prognostic feature. However, another study focused exclusively on CDKN2A/INK4A deletion failed to show an association with prognosis. A formal meta-analysis of this issue concluded that CDKN2A/INK4A aberration is a strong negative prognostic factor.
A number of other molecular markers have been proposed to have prognostic impact in Ewing sarcoma. In one study telomerase activity in the peripheral blood during follow-up, but not at initial diagnosis, was strongly associated with disease recurrence. None of the patients in this study with low telomerase activity during follow-up had disease recurrence. CD56 co-expression has been reported to be a favorable prognostic indicator in one series. One report suggested that serum IGF-1R levels were prognostic, although two other studies found no prognostic impact of IGF-1R levels. Two studies identified expression of the genes encoding glutathione S-transferases, enzymes involved in chemotherapy metabolism, as novel prognostic factors.
Treatment of Patients with Nonmetastatic Ewing Sarcoma
Local Control
Patients with nonmetastatic Ewing sarcoma require aggressive multimodality therapy to achieve long-term disease control. Local control of the primary tumor plays a key role in modern treatment for these patients. Historically, patients received radiation therapy alone as local control. Surgical local control was less commonly used because therapy was generally viewed as palliative. With improved outcomes thanks to chemotherapy, however, the late effects of radiotherapy on long-term survivors of Ewing sarcoma became apparent. In addition, limb-salvage surgical techniques became more advanced. As a result, surgical local control of resectable tumors has become the standard of care. Radiotherapy for local control of the primary tumor is now generally reserved for unresectable tumors or tumors that have been resected with inadequate margins. This section reviews the principles of radiotherapy and surgical resection in the care of patients with Ewing sarcoma.
Surgical resection is generally recommended for those tumors deemed completely resectable with wide or radical margins. For extremity tumors, surgical resection may take the form of an amputation or a limb-sparing procedure. Rotationplasty is another option for patients with femoral tumors. Technical advances, such as expandable implanted prostheses, have made limb-sparing procedures increasingly appealing. However, the choice of a limb-sparing procedure over other surgical options must not compromise the adequacy of the resection. Tumor infiltration of surrounding neurovascular structures, muscle, or skin should raise concern that a limb-sparing procedure will not result in the desired wide resection. Often neoadjuvant chemotherapy will produce sufficient response to allow for limb salvage when this option did not appear feasible at initial presentation. In a cooperative group analysis of chest wall tumors, resection after neoadjuvant chemotherapy was associated with higher rates of negative margins compared to initial resection. However, data from a single institution suggested that neither the likelihood of an adequate margin nor the overall outcome differs between patients who had surgical resection at initial diagnosis compared with patients who had surgical resection after a period of neoadjuvant chemotherapy. Nevertheless, most available data support the current standard approach of neoadjuvant chemotherapy before planned surgical resection.
Investigators from the CESS group and the Rizzoli Institute evaluated the impact of surgical margins on outcome in Ewing sarcoma in two retrospective case series. These groups both reported the rate of obtaining an adequate surgical margin as approximately 75%, among those patients selected for planned definitive resection. Both groups observed that an adequate margin is more difficult to obtain in axial tumors. For example, the Rizzoli group reported that approximately one quarter of the extremity tumors treated with a limb-salvage procedure had inadequate margins. In contrast, none of the paraspinal or sacral tumors in their large series of surgically treated patients had an adequate margin. The Rizzoli group observed that the 5-year event-free survival rate was higher for patients with an adequate surgical margin, although this finding may reflect improved outcome with smaller, more resectable tumors. In both series a small group of patients with inadequate surgical margins did not receive postoperative radiotherapy. Only 14% to 21% of these patients experienced a local recurrence. However, the risk of local recurrence was diminished by the addition of postoperative radiotherapy, which is the current standard practice.
Radiation therapy alone can provide adequate local control in Ewing sarcoma. In fact, the radiosensitivity of these tumors was one of the early characteristics that distinguished Ewing sarcoma from osteosarcoma. Local control rates with definitive radiation therapy in large series have been reported to range from 53% to 86%. For patients treated with radiation therapy alone, axial tumors (particularly pelvic tumors) have a higher rate of local failure than extremity tumors. One group reported that patients with metastatic disease treated with radiation alone or surgery plus radiation have a higher rate of local failure compared with patients who have nonmetastatic disease.
Several studies have evaluated the appropriate treatment volume, dose, timing, and schedule of radiotherapy in Ewing sarcoma. The IESS-1 study group reported on their experience with radiotherapy. All patients received radiotherapy as the mode of local control in this study. Across the range of doses administered (30 Gy to more than 60 Gy), no differences in local failure rates were observed, although very few patients received less than 40 Gy. Most patients received radiation to the whole bone involved with tumor, but some patients were treated with more limited fields. Patients who received radiotherapy to the primary tumor plus a 5-cm margin had the same local failure rate as patients treated with whole bone radiotherapy (8% for both groups). Patients treated with less than a 5-cm margin had an inferior local control rate.
The Pediatric Oncology Group conducted a study from 1983 and 1988 with the specific aim of determining the appropriate radiation volume for patients with Ewing sarcoma of the bone. Patients with tumors in expendable bones were recommended for surgical resection. Other patients were randomized to receive whole bone irradiation or involved field radiation. Patients treated with whole bone irradiation received 39.6 Gy to the entire bone containing tumor plus a boost to 55.8 Gy to the tumor with a 2-cm margin. Patients treated with involved field radiation received 55.8 Gy to the tumor with a 2-cm margin. Because of enrollment issues, 20 patients were randomized to whole bone radiation and 20 patients to involved field radiation. An additional 54 patients were nonrandomly assigned to involved field radiation. The 5-year event-free survival rate did not differ between patients randomized to whole bone radiation and patients randomized to involved field radiation (37% and 39%, respectively). The local control rate was 53% in both treatment arms. Of note, patients who received radiotherapy according to protocol guidelines for dose and volume had a superior local control rate (80%) compared with patients with major deviations (16% local control rate) or minor deviations (48% local control rate). Deviations in treatment volume seemed to be more critical. These results indicate that involved field radiation is an acceptable mode of local control for these patients but that treatment to the initial tumor volume plus an adequate margin is necessary.
Several retrospective single-institution case series have evaluated the impact of radiation dose on local control in Ewing sarcoma. In one series patients treated with less than 49 Gy had a 5-year local control rate of 37% compared with 89% for patients treated with 49 Gy or more. In this study higher doses seemed particularly important for larger tumors. In another series patients with larger tumors had a uniform local failure rate regardless of radiation dose, whereas patients with smaller tumors treated with more than 40 Gy had a higher local control rate compared with patients who had smaller tumors treated with less than 40 Gy. A third case series reported no difference in outcome between patients treated with radiation doses above and below 54 Gy. The CESS-81 trial randomized extremity tumors to receive 46 Gy or 60 Gy. The local failure rate was similar in these two groups.
Three studies have specifically commented on the use of lower-dose radiotherapy. The first study administered approximately 30 Gy in patients mainly in conjunction with some type of surgical resection, although five patients received radiotherapy only. No local failures were noted in this group. The other two studies administered 30 to 36 Gy as definitive local control to patients with an objective response to neoadjuvant chemotherapy. This strategy resulted in an unacceptably high local failure rate.
The timing of radiotherapy may be important for patients receiving definitive radiotherapy and not for patients receiving radiotherapy after surgery. A retrospective analysis of patients treated with surgery followed by radiation therapy in the CESS-86 and EICESS-92 trials demonstrated that the interval between surgery and radiation did not affect the local control rate or event-free survival rate, even up to intervals beyond 90 days. In contrast, a retrospective analysis of patients with pelvic Ewing sarcoma suggested that initiating definitive radiotherapy earlier may result in a lower local failure rate. A pooled analysis has also suggested that longer periods (15-18 weeks) of neoadjuvant chemotherapy before starting radiation therapy may have a negative impact on the overall survival rate.
Two retrospective case series evaluated hyperfractionated radiotherapy in the management of Ewing sarcoma. Both series concluded that local control rates were not improved by hyperfractionation. One group reported better functional results with the hyperfractionated schedule. The CESS-86 trial randomized patients to conventional or hyperfractionated radiation schedules. Patients on the conventional schedule had chemotherapy held during radiotherapy, while patients on the hyperfractionated schedule continued to receive chemotherapy during radiation. For patients treated with radiotherapy alone, the local control rate was 82% for patients on the conventional schedule and 86% for patients on the hyperfractionated schedule. The radiation schedule did not affect overall or relapse-free survival rates. The radiation schedule did not affect disease control in patients treated with surgery followed by radiation.
On the basis of this experience with radiotherapy for Ewing sarcoma, current practice is for patients with unresectable tumors to receive definitive radiotherapy after 4 to 6 cycles of chemotherapy. Most groups administer 54.4 to 60 Gy as definitive radiotherapy. Patients typically receive treatment as 45 Gy to the pretreatment volume plus a safety margin of at least 2 cm followed by a boost to full-dose radiotherapy to the treatment volume remaining after neoadjuvant chemotherapy. Tumors with soft tissue extension, but not infiltration, into the chest or pelvic cavities often receive treatment to the postchemotherapy volume plus a safety margin. Some centers advise full-dose preoperative radiotherapy to tumors that may become fully resectable with such therapy. Patients with inadequate surgical margins are recommended to receive postoperative radiation. The EURO-EWING group also recommends postoperative radiotherapy for patients with adequate surgical margins and a poor histologic response to chemotherapy, although this practice is not standard in North America. The EURO-EWING group recommends a hyperfractionated schedule of radiotherapy, whereas the standard in North America remains conventional fractionation.
Several groups have compared the three available modes of local control: surgery, definitive radiation, and surgery plus radiation. Several studies have clearly documented that patients who undergo surgical resection have higher local control rates or improved overall outcomes (or both) compared with patients who receive definitive radiotherapy only. * These studies must be interpreted with caution because of the presence of confounding by indication. For example, because small extremity tumors are typically selected for surgery and large axial tumors are typically managed with radiotherapy alone, the outcome between these two groups might be expected to differ on account of reasons other than the chosen mode of local control. Indeed, not all studies have concluded that surgery provides superior disease control compared with radiation. A retrospective single-institution study of 76 patients with nonmetastatic Ewing sarcoma concluded that the overall survival rate and the rate of local control were equivalent between patients who received local control as surgery, radiation, or surgery plus radiation, although the power to detect a difference may have been limited by the small size of this study. A secondary analysis of data from the United Kingdom Children’s Cancer Study Group (UKCCSG) ET-2 trial reached the same conclusion. In addition, the overall rate of disease recurrence did not differ between local control groups. In the SE 91-CNR Italian cooperative group trial, the 3-year event-free survival rates did not differ between patients who received surgery alone and those who received radiotherapy alone.
* References .
Although a randomized trial might resolve the issue of optimal mode of local control, such a randomization would be untenable to patients. Instead, several analyses have evaluated this issue by attempting to control for the known confounding factors influencing both outcome and choice of local control modality. An analysis from the Rizzoli Institute showed that patients treated with definitive radiotherapy had inferior event-free survival rates compared with patients who received surgery alone, even after controlling for age, tumor size, and serum LDH level. This group did not control for tumor site in this analysis. Two similar previous analyses by this same group controlled for tumor site, among other factors. In these analyses patients treated with radiotherapy did not have an increased risk of relapse. The EICESS/CESS experience with local control has also been retrospectively reviewed in several analyses. On univariate analyses, these studies have demonstrated that patients treated surgically have improved local control compared with patients treated with definitive radiotherapy. Although this group has not reported a multivariate analysis of outcome based on local control, they have reported a subgroup analysis based on tumor size (above and below 100 cm 3 ) and site (axial versus appendicular). Patients treated with radiation alone had inferior local control for all combinations of tumor size and site except small central tumors. However, patients treated with surgery appeared to have a higher rate of systemic failure compared with patients treated with radiation alone, such that the overall rate of disease recurrence was the same between groups. This finding has not been reported by other groups. An analysis of 220 patients treated at St. Jude Children’s Research Hospital also demonstrated that surgical resection was associated with lower rates of local failure, even after controlling for tumor size and year of treatment. This analysis did not control for tumor site, however. Significantly, this analysis and a prospective randomized trial demonstrated lower rates of local failure in patients treated with more modern chemotherapy regimens that included ifosfamide and etoposide.Systemic Treatment with Chemotherapy
The use of chemotherapy for patients with Ewing sarcoma has revolutionized the care of these patients. Before the 1970s, standard therapy for this disease consisted solely of local control directed at the primary tumor. Most patients treated in this manner ultimately died from local or systemic recurrent disease. In one series patients treated with local control alone had a median survival rate of 11 months. In the 1960s and 1970s, evidence emerged that systemic chemotherapy could prevent recurrent disease in a substantial number of patients with Ewing sarcoma. This section reviews the development of modern chemotherapy regimens for this disease. Table 61-3 lists major chemotherapy regimens evaluated for Ewing sarcoma along with abbreviations for these regimens. Table 61-4 summarizes the major cooperative group trials that have contributed to current treatment approaches to this disease.
| Abbreviation | Regimen |
|---|---|
| VAC | Vincristine, actinomycin-D, and cyclophosphamide |
| VACA | Vincristine, actinomycin-D, cyclophosphamide, and doxorubicin |
| VDC | Vincristine, doxorubicin, and cyclophosphamide |
| IE | Ifosfamide and etoposide |
| VAI | Vincristine, actinomycin-D, and ifosfamide |
| VDI | Vincristine, doxorubicin, and ifosfamide |
| VAIA | Vincristine, actinomycin-D, ifosfamide, and doxorubicin |
| VIDE | Vincristine, ifosfamide, doxorubicin, and etoposide |
| Trial | Years | Regimen(s) | 5-year EFS or RFS | Reference(s) |
|---|---|---|---|---|
| VACA Trials | ||||
| IESS-1 | 1973-1978 | VACA VAC + WLI VAC | 60% 44% 24% | |
| IESS-2 | 1978-1982 | High dose VACA Protracted VACA | 73% nonpelvic 55% pelvic 56% nonpelvic | |
| First French | 1978-1984 | VACA | 52% (4 year) | |
| UKCCSG ET-1 | 1978-1986 | VACA | 41% | |
| CESS-81 | 1981-1985 | VACA | 55% | |
| French EW88 | 1988-1991 | Protracted VACA | 58% | |
| VACA/IE Trials | ||||
| Second French | 1984-1987 | VAI/VDI | 52% | |
| CESS-86 | 1986-1991 | VAIA for high risk VACA for low risk | 53% (10 year) 49% (10 year) | |
| UKCCSG ET-2 | 1987-1993 | VAI/VDI | 62% | |
| INT-0091 | 1988-1992 | VACA/IE VACA | 69% 54% | |
| SSG IX | 1990-1996 | VDI/PDI | 58% | |
| Italian SE 91-CNR French EW-93 | 1991-1997 1993-1999 | VACA/IE VACA for low risk VACA/IE for intermediate risk | 69% 70% 54% | |
| EICESS-92 | 1992-1999 | VAIA for low risk VACA for low risk VAIA for high risk VAIAE for high risk | 68% 67% 44% 52% | |
| Dose Intensified VACA/IE Trials | ||||
| INT-0154 | 1995-1998 | VACA/IE standard VACA/IE intensified | 72% 70% | |
| AEWS0031 | 2001-2005 | VACA/IE 3-week VACA/IE 2-week | 65% 73% | |
| EURO-EWING 99 | Ongoing | VIDE | Ongoing | |
| AEWS1031 | Ongoing | VDC/IE 2-week VDC/IE/TC 2-week | Ongoing | |
Early Improvements with Vincristine, Actinomycin-D, Doxorubicin, and Cyclophosphamide
Given the poor outcomes observed with local control alone, investigators began using single-agent chemotherapy for Ewing sarcoma. Although single-agent therapy did not substantially improve the long-term outcomes for these patients, these studies did identify a group of drugs capable of inducing tumor regression. Some of the most effective agents included vincristine, actinomycin-D, doxorubicin, and cyclophosphamide. Very quickly, investigators began to evaluate the use of these drugs in combination for patients with Ewing sarcoma.
One group evaluated two different combinations: vincristine with cyclophosphamide given in 8-week cycles for five cycles and vincristine, actinomycin-D, doxorubicin, and cyclophosphamide (VACA) given in weekly components for 18 months. Most patients received radiotherapy as the mode of local control. In both regimens chemotherapy and radiotherapy started simultaneously at the initiation of treatment. Patients who received either regimen had a statistically significant improvement in outcome compared with historical controls who received only local control, suggesting that the VACA regimen resulted in better outcomes than the vincristine-cyclophosphamide regimen.
Another group reported on 30 patients with nonmetastatic Ewing sarcoma treated with 18 months of vincristine, doxorubicin, cyclophosphamide, and procarbazine. Chemotherapy and radiotherapy were given concurrently at the start of therapy. The 6-year disease-free survival rate was 49%, which was a substantial improvement on outcomes in historical controls treated with radiotherapy alone.
The Memorial Sloan Kettering Cancer Center treated 20 patients with localized Ewing sarcoma with 18 to 20 months of VACA as adjuvant therapy. The cumulative dose of doxorubicin used in this study typically exceeded 500 mg/m 2 , higher than in most other studies. The 5-year disease-free survival rate was 75%, perhaps highlighting the sensitivity of Ewing sarcoma to doxorubicin. The cardiotoxicity associated with these doses of doxorubicin resulted in a reduction in the cumulative doxorubicin exposure in subsequent studies by this group.
The St. Jude Children’s Research Hospital evaluated the use of neoadjuvant therapy with cyclophosphamide and doxorubicin for five cycles. An early report of this regimen reported that 19 of 23 evaluable patients had a complete response to therapy. After these five cycles of neoadjuvant therapy, patients received local control, usually with radiotherapy. With the start of local control, patients began adjuvant therapy with vincristine and actinomycin-D for approximately 11 weeks followed by an additional six cycles of cyclophosphamide and doxorubicin. At the time of a follow-up report of 50 evaluable patients with nonmetastatic disease treated in this manner, 17 patients had developed recurrent disease. A larger cooperative group study evaluating radiotherapy in Ewing sarcoma used a similar chemotherapy regimen and reported a 5-year event-free survival rate of 51% in patients with nonmetastatic disease.
The Rizzoli Institute performed a series of trials evaluating both adjuvant (after local control) and neoadjuvant (before local control) VACA regimens. In the first trial patients received adjuvant therapy with vincristine, doxorubicin, and cyclophosphamide (VDC) for 2 years. At the time of diagnosis, patients began chemotherapy in concert with local control of the primary tumor. In the 85 patients with localized disease treated in this manner, the 5-year event-free survival rate was 34%. In contrast, only 5% of historical controls treated with local control only remained continuously free of disease. The successor study treated 59 patients with localized disease with 18 months of VACA. In this study the 5-year event-free survival rate was 59%, suggesting a benefit to the addition of actinomycin-D. The next study by this group evaluated VDC given as three neoadjuvant cycles followed by VACA chemotherapy after local control for a total of 15 months. The 5-year event-free survival rate for the 108 patients with localized disease treated in this study was 49%. Compared to VACA given exclusively as adjuvant therapy, this result suggests that the change to neoadjuvant therapy did not improve outcomes for these patients.
Based on these promising results, several cooperative groups set out to evaluate vincristine, actinomycin-D, and cyclophosphamide (VAC) and VACA regimens in large groups of uniformly treated patients. IESS-1 was the first randomized study in patients with Ewing sarcoma. This study enrolled patients from 1973 and 1978. The 342 patients with nonmetastatic Ewing sarcoma were randomized to one of three treatment arms. Patients in the first and second treatment arms received VAC, with patients in the second treatment arm also receiving prophylactic whole lung irradiation. Patients in the third treatment arm received VAC plus doxorubicin. Patients in this arm received doxorubicin every 12 weeks and actinomycin-D every 12 weeks, such that they received one of these agents every 6 weeks. Patients in the VAC arms received actinomycin-D every 12 weeks. All patients received radiotherapy to the primary tumor. Patients who received doxorubicin had a 5-year relapse-free survival rate of 60% compared with only 24% for patients who received VAC only. Patients who received VAC with whole lung irradiation had an intermediate 5-year relapse-free survival rate of 44%. These results indicated that the addition of doxorubicin to VAC significantly improved outcomes compared with either VAC alone or VAC with whole lung irradiation. The contribution of increased dose intensity in the doxorubicin arm to this improved outcome is not clear. Subgroup analysis showed that outcomes were inferior for patients with pelvic primary tumors and did not differ by treatment arm for these patients. Doxorubicin performed as well as whole lung irradiation in preventing lung metastasis. Based on these results, four-drug therapy with VACA became the standard chemotherapy approach for patients with Ewing sarcoma.
The IESS-2 study expanded on these results by randomizing patients with nonmetastatic disease not involving the pelvis to two different VACA dosing schedules. Patients on one schedule received cycles of therapy every 3 weeks, and patients on the other schedule received more moderate doses of the same drugs at more frequent intervals. All patients received treatment for approximately 20 months. A total of 214 patients were enrolled in this study from 1978-1982. The 5-year relapse-free survival rate for patients with nonpelvic tumors who received higher-dose intermittent VACA was 73%, compared with 56% for patients who received more protracted VACA. Overall survival rate results showed a similar pattern. These results indicated a significant survival advantage for the higher-dose intermittent dosing schedule, at least for patients with tumors arising outside the pelvis.
The first German cooperative group trial for this disease (CESS-81) also used VACA administered in 3-week cycles. This trial enrolled 93 patients with nonmetastatic disease from 1981 to 1985. Treatment duration was only 10 months, which was shorter than treatment on the IESS protocols. The 5-year disease-free survival rate was 55%, including patients with pelvic tumors. This result is similar to the outcome of VACA-treated patients on IESS-1, which also included patients with pelvic tumors. This result suggested that a shorter treatment duration using higher-dose regimens every 3 weeks does not compromise outcome.
The first Ewing sarcoma study of the United Kingdom Children’s Cancer Study Group (UKCCSG ET-1) sought to standardize the treatment of newly diagnosed Ewing sarcoma using VACA-based chemotherapy. This nonrandomized trial enrolled 142 patients from 1978 to 1986. Patients received one or two induction cycles of chemotherapy with VDC. Local therapy, with a preference for definitive radiotherapy, occurred next while patients received weekly cyclophosphamide and vincristine. Patients then received cycles of VDC alternating every 3 weeks with VAC for a total of 1 year of therapy. The 5-year relapse-free survival rate was 41%, and the 5-year overall survival rate was 44% for patients with nonmetastatic disease. The study authors suggest that the inferior outcome in this trial compared with that of other VACA-based trials at the time may have been due in part to decreased early doxorubicin dose intensity.
The first French cooperative group study of Ewing sarcoma also used VACA-based chemotherapy as well as radiotherapy as the preferred mode of local control. This study enrolled 95 patients between 1978 and 1984. Patients received cycles of VDC alternating with VAC for a total of 16 months of treatment. The 4-year disease-free survival rate for patients with nonmetastatic disease was 52%.
A follow-up French cooperative group study (EW88) enrolled 141 patients with nonmetastatic disease from 1988 to 1991. This study administered cyclophosphamide in a more protracted manner, following a regimen piloted at St. Jude’s Children’s Research Hospital in the 1970s. Patients received initial chemotherapy with cyclophosphamide given daily for 7 days followed by doxorubicin on day 8. Patients received five cycles of this therapy before proceeding to local control. Surgery was recommended when possible. Radiotherapy was reserved for patients with unresectable tumors, incompletely resected tumors, or tumors with more than 5% viable tumor after initial chemotherapy. Following local control, patients received vincristine and actinomycin-D for 12 weeks followed by six additional cycles of cyclophosphamide and doxorubicin using the same schedule used neoadjuvantly. Administering these standard four drugs in this manner resulted in a 5-year disease-free survival rate of 58%.
Addition of Ifosfamide and Ifosfamide with Etoposide to VACA-Based Therapy
An initial report from the U.S. National Cancer Institute (NCI) demonstrated a response rate of 45% in 20 patients with recurrent Ewing sarcoma treated with a 5-day course of ifosfamide. A follow-up study by this group evaluated ifosfamide combined with etoposide (IE) for patients with recurrent pediatric tumors. Of the 17 evaluable patients with recurrent Ewing sarcoma included in the preliminary report, 16 patients had a partial response to therapy, for a response rate of 94%. A larger cooperative group study sponsored by the Pediatric Oncology Group included 55 patients with recurrent Ewing sarcoma treated with ifosfamide and etoposide. The response rate in this study was 25%. Based on these promising results in patients with recurrent disease, the St. Jude Children’s Research Hospital evaluated IE in patients with newly diagnosed high-risk disease. This group gave three cycles of IE as an initial window before proceeding with doxorubicin and cyclophosphamide chemotherapy. Only 1 of 26 patients failed to respond to IE, for a response rate of 95%.
A number of single-institution studies incorporated ifosfamide and etoposide into the VACA backbone of therapy for newly diagnosed patients. One study from the NCI administered IE alternating with VDC or VAC for a total of 18 3-week cycles. Ifosfamide was administered both before and after local control. A total of 31 patients with nonmetastatic disease were treated and had a 5-year event-free survival rate of 64%. This outcome was significantly better than historical controls with nonmetastatic disease treated with VDC and total body irradiation with stem cell rescue. Two studies from the Rizzoli Institute introduced ifosfamide at different times and reached different conclusions. In the REN-2 study, patients with localized disease received ifosfamide for the first time after local control. The cumulative ifosfamide dose was 54 grams/m 2 . The 5-year event-free survival rate for the 82 patients treated was 51%. This result was not different from historical controls treated with VACA-based therapy. In the REN-3 study, ifosfamide was introduced before local control with the cumulative dose held constant at 54 grams/m 2 . The 157 patients with nonmetastatic disease had a 5-year event-free survival rate of 71%, which was significantly better than any of the previous chemotherapy regimens evaluated at their center. These results suggest that early introduction of ifosfamide may improve outcome.
Several cooperative group studies have evaluated the efficacy of ifosfamide both with and without etoposide. One of the first cooperative group studies incorporating ifosfamide was the second French cooperative group study of Ewing sarcoma. Patients enrolled from 1984 to 1987 and received initial therapy with alternating 3-week cycles of vincristine, actinomycin-D, and ifosfamide (VAI) and vincristine, doxorubicin, and ifosfamide (VDI) for a total of six cycles. Patients then underwent local control, with a preference for complete surgical resection. All patients were to receive radiotherapy, with the dose dependent on the extent of surgical resection. During radiotherapy patients received two cycles of vincristine with doxorubicin and two cycles of vincristine with ifosfamide. Following local control, patients received alternating 3-week cycles of VAI and vincristine with doxorubicin to complete a total of 1 year of therapy. The 5-year disease-free survival rate was 52% for 95 patients with nonmetastatic disease. This result prompted this group to conclude that substituting ifosfamide for cyclophosphamide did not improve outcomes. In addition, patients in this study had a higher than expected incidence of diminished cardiac function, prompting early closure of the study.
The CESS-86 trial was the first cooperative group attempt to risk-stratify patients and alter therapy based on risk. Patients with small (less than 100 mL tumor volume) extremity tumors received VACA given as 3-week cycles for 36 weeks. Patients with large (100 mL or greater) tumors or axial tumors received identical therapy except ifosfamide was substituted for cyclophosphamide (VAIA). For the 301 nonmetastatic patients treated on this study, the ten-year event-free survival did not differ between patients who received VACA or VAIA (49% and 53%, respectively). However, multivariate analysis demonstrated that, controlling for tumor size and site, VACA chemotherapy was less effective than VAIA.
The French EW-93 study prescribed doxorubicin and cyclophosphamide for all patients with localized disease and reserved IE for patients with a poor response to three cycles of that therapy. Patients with less than 50% regression of soft tissue mass on first evaluation were nonrandomly assigned to receive two cycles of IE before surgical resection. Of 214 patients, 33 received IE, and only eight of these refractory patients responded to IE before local control measures.
The UKCCSG ET-2 study evaluated the substitution of ifosfamide for cyclophosphamide in newly diagnosed patients. This nonrandomized trial enrolled 243 patients from 1987 to 1993. Patients received VDI for four 3-week cycles before undergoing local control. In contrast to ET-1, ET-2 suggested a preference for surgery as mode of local control. Following local control, patients received VAI to complete 1 year of therapy. Patients received a cumulative doxorubicin dose of 420 mg/m 2 and cumulative ifosfamide dose of 114 grams/m 2 . The 5-year relapse-free survival rate was 62% for patients with nonmetastatic disease. These results demonstrate a substantial improvement over the results of the UKCCSG ET-1 study. These improvements may be due to increased doxorubicin dose intensity, use of ifosfamide, preference for surgery as local control, or a combination of these factors.
An Italian cooperative group study (SE 91-CNR) enrolled 160 evaluable patients with nonmetastatic Ewing sarcoma from 1991 to 1997. Patients treated on this protocol received initial therapy with alternating 3-week cycles of VDC and VAI. After 24 weeks of therapy, patients received alternating 3-week cycles of IE and VAC to complete 36 total weeks of therapy. Surgery was the preferred mode of local control for resectable tumors. The 5-year event-free survival rate was 69.4%.
A Scandinavian Sarcoma Group trial (SSG IX) enrolled patients with metastatic and nonmetastatic Ewing sarcoma from 1990 to 1996. All patients received 35 weeks of chemotherapy given as 3-week cycles. Patients received cycles of VDI every 3 weeks, with cisplatin, doxorubicin, and ifosfamide substituted at cycles 2, 5, 8, and 11. Surgery was the preferred mode of local control. The 5-year metastasis-free survival rate was 58% for the 73 patients with initially nonmetastatic disease. The 5-year overall survival rate for these patients was 70%. The 5-year overall survival rate for patients presenting with metastatic disease was 28%. These results compare favorably to those from other contemporary trials.
Two randomized cooperative group trials have directly compared VACA-based chemotherapy to ifosfamide-containing chemotherapy. The North American intergroup study INT-0091 enrolled 518 patients with metastatic and nonmetastatic Ewing sarcoma from 1988 to 1992. The standard arm on this study consisted of 3-week cycles of VDC. The experimental arm consisted of 3-week cycles of VDC alternating with IE. In both treatment arms, actinomycin-D was substituted for doxorubicin after a cumulative doxorubicin dose of 375 mg/m 2 . All patients received 17 cycles of therapy, for a total study duration of 49 weeks. Local control occurred at week 12 of the therapy, with mode of local control determined on an individualized basis. The addition of IE significantly improved the outcome for patients with nonmetastatic disease, with a 5-year event-free survival rate of 69% compared with 54% for patients receiving VDC only. The incidence of local failure was also lower for patients who received IE, indicating that improved systemic therapy also improves local control. Based on these results, the combination of VDC and IE has become the standard therapy for patients with nonmetastatic Ewing sarcoma treated in North America.
The EICESS-92 study represents a collaborative effort between the UKCCSG and German cooperative groups. This trial enrolled 647 patients with either localized and metastatic Ewing sarcoma from 1992 to 1999. Patients with small (less than 100 mL tumor volume) localized tumors were deemed standard risk. These patients received initial chemotherapy with VAIA given as 3-week cycles followed by a randomization between continued VAIA or VACA. All other patients were deemed high risk and were randomized to receive VAIA or VAIA plus etoposide. Among standard-risk patients there were no statistically significant differences in event-free or overall survival rates between the randomized arms, although it is important to note that all patients received ifosfamide before randomization. Among high-risk patients there were trends suggesting improved outcomes with the addition of etoposide to VAIA, although no statistically significant differences were found.
Dose-Intensified VACA/IE Regimens
The next generation of chemotherapy regimens emphasizes increasing the dose intensity of known active agents in this disease. The P6 protocol from the Memorial Sloan Kettering Cancer Center used an augmented dose of cyclophosphamide given in combination with vincristine and standard-dose doxorubicin. Patients received a total of seven cycles of alternating VDC and IE along with local control of the primary tumor. The 4-year event-free survival rate for the 44 patients with localized disease was 82%, suggesting an improvement in outcome with higher-dose cyclophosphamide.
Two recent North American cooperative group studies have investigated strategies for dose-intensifying therapy. The first trial, known as INT-0154, included 478 eligible patients with nonmetastatic Ewing sarcoma diagnosed from 1995 to 1998. Patients were randomized at study entry to receive alternating 3-week cycles of VDC and IE using either a dose-intensified or standard-dose approach. The standard-dose arm emulated the therapy given to patients assigned to the experimental arm of INT-0091. Patients in the dose-intensified arm received higher individual doses of the alkylating agents than patients in the standard-dose arm, but cumulative drug doses were approximately equivalent between arms. Patients in the dose-intensified arm completed therapy in 30 weeks, whereas patients in the standard-dose arm completed therapy in 48 weeks. Dose-intensified treatment did not improve event-free or overall survival rates and was associated with increased toxicity, such that this approach is not recommended for this population.
In Children’s Oncology Group (COG) protocol AEWS0031, 568 patients with nonmetastatic Ewing sarcoma were randomized at study entry to receive cycles of standard-dose VDC and IE alternating every 3 weeks or every 2 weeks. Individual and cumulative drug doses were equivalent between treatment arms. All patients received 14 cycles of therapy, requiring 42 weeks in standard arm and 28 weeks in the interval compression arm. Interval compression was feasible, with a median cycle length of 15 days in the interval compression arm. Patients randomized to the interval compression arm had a superior event-free survival rate compared with patients randomized to the standard arm (73% versus 65% at 5 years). Toxicity was equivalent between both arms. Based on these findings, this approach has been adopted in North America as a standard practice for patients with localized Ewing sarcoma.
Several groups have evaluated chemotherapy regimens that combine multiple active agents given together during a cycle of therapy. In a protocol at St. Jude Children’s Research Hospital, 51 patients with Ewing sarcoma received ifosfamide, etoposide, cyclophosphamide, and doxorubicin given together for three cycles before local control. Patients then received alternating cycles of IE and cyclophosphamide with doxorubicin for a total of 41 weeks of therapy. The doses of ifosfamide, etoposide, and cyclophosphamide given after local control were higher than in previous studies. Patients were able to complete the planned neoadjuvant therapy, but myelosuppression limited the ability of patients to receive the planned adjuvant therapy on schedule. Specifically, only 66% of patients received all planned chemotherapy, and only 25% of patients received all planned chemotherapy on schedule. Despite difficulties in achieving the desired degree of dose intensification, outcomes remained favorable. The 3-year event-free survival rate for patients with localized disease was 78%.
A British group has piloted the use of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) as neoadjuvant therapy for this disease. In this study 30 patients with newly diagnosed Ewing sarcoma were treated with six 3-week cycles of VIDE. Because of hematologic toxicity, etoposide was dose reduced or omitted from 50% of the planned cycles. All attempts at collecting peripheral blood stem cells after a cycle of VIDE were successful. Of the 24 patients evaluable for radiographic response, only one patient had progressive disease. Patients in this pilot study received additional therapy following local control. The final results of this study are not yet available.
The European cooperative groups have initiated a combined trial known as EURO-EWING 99 that relies on VIDE as neoadjuvant therapy. All patients receive initial treatment with six 3-week cycles of VIDE. This trial then tailors therapy according to risk. Patients with small (less than 200 mL tumor volume) nonmetastatic tumors with good chemotherapy response are randomized to receive additional therapy with seven cycles of VAC or VAI. Patients with large tumors, pulmonary metastases, or poor chemotherapy response are randomized to receive additional therapy with seven cycles of VAI or high-dose therapy with stem cell rescue (discussed later). This randomized trial is ongoing.
Two nonrandomized trials have reported on the use of high-dose chemotherapy as a consolidation strategy for patients with localized Ewing sarcoma and adverse prognostic factors. In the French EW93 trial, patients with localized Ewing sarcoma were considered high risk if they had either a 30% or greater viable tumor at time of surgical resection or a 50% or less reduction in tumor volume by imaging for patients not candidates for surgical resection. These patients were nonrandomly assigned to receive high-dose chemotherapy with busulfan and melphalan. There were 48 patients who were classified as high risk, 34 of whom received high-dose chemotherapy. For the entire cohort of patients with high-risk disease, the 5-year event-free survival rate was 48%. A joint Italian-Scandinavian protocol adopted a similar approach, with busulfan and melphalan high-dose chemotherapy prescribed for patients with localized disease and macroscopic foci of tumor at the time of surgery or incomplete regression of soft tissue masses in patients not candidates for surgical resection. Outcomes for the 126 patients with these poor risk features who received high-dose therapy were similar to those of patients without poor risk features who did not receive high-dose therapy. Given the nonrandomized study designs and risk of selection bias for patients who proceeded to high-dose therapy in these two trials, the role of this approach in patients with localized Ewing sarcoma remains unclear.
Management of Tumors Arising at Special Sites
Pelvic Ewing Sarcoma
Ewing sarcoma frequently arises in the pelvis. These pelvic tumors tend be large and associated with crucial viscera, nerves, and bones (particularly of the acetabulum), making complete surgical resection difficult. The local failure rate for patients with pelvic primary tumors ranges from 15% to 27%, which is higher than the local failure rate typically reported in appendicular tumors. In one large series, even after exclusion of patients with a local relapse, the outcome for patients with pelvic primary tumors was still inferior to outcomes at other sites. This result suggests that both systemic control and local control are inadequate for these patients.
Recognizing the poor outcome of patients with pelvic primary tumors, the IESS-2 trial nonrandomly assigned patients to the higher-dose intermittently scheduled VACA arm of that study. In addition, an increased emphasis was placed on surgical control of pelvic tumors. With this strategy, patients with pelvic tumors had a 5-year relapse-free survival rate of 55% compared with only 23% for patients in the IESS-1 trial. Because the higher dose VACA arm resulted in better outcomes in the randomized nonpelvic patients treated in IESS-2, the improvements in outcome seen in patients with pelvic tumors may have been attributable to better systemic control, better local control, or both. Indeed, patients with pelvic tumors in IESS-2 were more likely to undergo complete surgical resection and had a lower local failure rate compared to patients with pelvic tumors in IESS-1. Compared with patients who did not have a complete resection, complete resection did not improve the overall or relapse-free survival rate in these patients in IESS-2.
The optimal mode of local control for pelvic tumors has been difficult to determine with rigorous studies. A report from the Mayo Clinic found that patients with pelvic Ewing sarcoma treated with surgery had a statistically significant superior overall survival rate compared with patients treated without surgery. In several other series of patients with pelvic primary tumors, the mode of local control has not clearly been shown to affect outcome, although some trends have suggested improvements in survival rates among patients treated surgically. Some groups have also reported a trend toward a lower local recurrence rate in patients treated surgically, although not all groups have reported this trend. The results of these studies are often difficult to evaluate insofar as patients treated with complete surgical resection typically have small resectable tumors. However, two groups have attempted to control for this and other confounding factors. The French cooperative group reported on 53 patients with pelvic Ewing sarcoma. After controlling for size and other potential confounding factors, researchers found that patients treated with radiotherapy alone had an increased risk of recurrence and death compared with patients who received surgery or surgery with radiotherapy. The COG retrospectively analyzed 75 patients with pelvic primary tumors treated on INT-0091. In contrast to the results of the French group, the mode of local control did not affect event-free survival or local failure rates after controlling for confounding factors such as size. As observed in the main analysis of all tumor sites, patients with pelvic tumors who were randomized to receive VACA plus IE on this study had a lower risk of local failure compared with patients randomized to receive VACA alone. Given this uncertainty and the risk of second tumors from radiotherapy, many groups choose to resect pelvic tumors that can be resected without functional consequences. For tumors that cannot be resected without functional consequences, options include either definitive radiotherapy alone or preoperative radiotherapy with the possible addition of wide resection.
Chest Wall Ewing Sarcoma
Chest wall tumors also require specialized management, particularly insofar as these tumors often invade the pleura or have associated pleural effusions. Some data suggest that surgical resection of chest wall tumors provides superior local control, although these analyses do not account for differences in tumor size that influence resectability. Indeed, other analyses have not shown a difference in outcome based on mode of local control for chest wall tumors. Results from INT-0091 and INT-0154 indicate that neoadjuvant chemotherapy before attempted resection of a chest wall tumor increases the likelihood of obtaining negative margins compared with results from attempted resection before chemotherapy. Delayed resection at this site therefore decreases the need for postoperative radiotherapy in these patients. Avoiding chest wall radiation is particularly appealing given the added pulmonary and cardiac toxicities associated with this therapy.
Whereas several reports have suggested that an associated pleural effusion did not worsen outcome, patients with chest wall tumors and pleural effusions were often recommended to receive hemithorax irradiation in these studies. Indeed, a report from the French cooperative group suggested that patients with rib primary tumors and an associated pleural effusion had an increased risk of local relapse without radiotherapy. In the EICESS/CESS studies, patients who received hemithorax irradiation had more unfavorable features than patients who did not receive hemithorax irradiation. Despite this difference, the rate of local relapse was the same in both groups, whereas the rate of systemic relapse was higher in the group of patients who did not receive hemithorax irradiation. This result may be interpreted as evidence of the direct benefit of hemithorax irradiation in preventing dissemination of residual tumor cells from the pleural space. Alternatively, hemithorax irradiation may reduce the volume of lung at risk for subsequent relapse, because the IESS-1 researchers noted that whole lung irradiation can prevent lung metastasis in patients with initially nonmetastatic disease. Patients with chest wall primary tumors and positive pleural fluid cytology or pleural-based nodules treated according to the most recent COG protocol received hemithorax radiation.
Ewing Sarcoma of the Spine
Management of patients with tumors arising along the spinal column requires careful planning by a multidisciplinary team, including oncologists, radiation oncologists, orthopedic surgeons, general surgeons, and neurosurgeons. Small case series have suggested that local control and overall outcomes for patients with primary tumors at this site do not differ from outcomes for patients with tumors arising from other sites. One group reported that sacral primary tumors fare less well than other vertebral tumors, although this result was not replicated in other series. The largest reported experience with vertebral Ewing sarcoma comes from the EICESS/CESS studies. A total of 116 patients had tumors arising from cervical, thoracic, or lumbar vertebrae. Given the difficulties in obtaining a complete resection at this site, all but four patients received radiotherapy as part of local control of the tumor. The local failure rate for patients treated with radiotherapy alone was not significantly different from that of patients treated with surgery plus radiotherapy (22.6% and 18.7%, respectively). Local relapses typically occurred within the radiation field, suggesting that inadequate selection of radiotherapy fields was not to blame for these local relapses. Despite the fact that the spinal cord limits radiotherapy dosing at this site, the local failure rate for patients with vertebral tumors treated with radiotherapy alone was not higher than the local failure rate for patients with tumors of any location treated with radiotherapy alone. This effect may result from the generally smaller size of paraspinal tumors compared with that of other tumors selected for local control with definitive radiotherapy. It should be noted, however, that patients with a vertebral tumor treated with radiotherapy who experience local recurrence often have few local control options because additional radiotherapy may compromise the spinal cord.
Soft Tissue Ewing Sarcoma
Historically, patients with soft tissue Ewing sarcoma were not included in cooperative group clinical trials for patients with Ewing sarcoma of bone. Instead, patients with soft tissue Ewing sarcoma have often been included in protocols designed for patients with other soft tissue sarcomas, including rhabdomyosarcoma. More recent work suggests that these patients should receive treatment following Ewing sarcoma protocols. For example, one study compared patients treated according to International Society of Paediatric Oncology (SIOP) protocol MMT89 without doxorubicin to patients treated on the French EW93 protocol with doxorubicin. Although it was a nonrandomized comparison, baseline patient characteristics were similar between both groups, but event-free and overall survival rates were superior for patients treated according to the EW93 protocol. In North American study INT-0154, patients with soft tissue disease had outcomes equivalent to those of patients with skeletal primary tumors. These results support the current practice of including these patients in modern trials for Ewing sarcoma.
Summary of Outcomes for Patients with Nonmetastatic Ewing Sarcoma
Patients with Ewing sarcoma have shown great improvements in outcome over the past 4 decades. For patients with localized tumors, expected survival rates have increased to greater than 70% with modern multimodality therapy. Figure 61-8 demonstrates these improvements graphically, using Surveillance, Epidemiology, and End Results (SEER) data from 1975 to 2000. A similar pattern of steady improvement over time has also been reported using data from a large European registry.
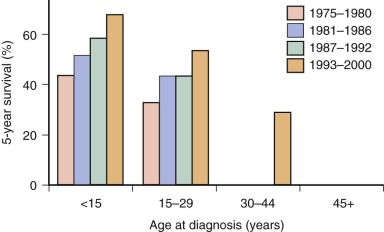
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


