Some diseases, such as myeloma with hypergammaglobulinemia or hyper-albuminemia due to severe dehydration, can cause an elevation in the serum total calcium level without any rise in serum ionized calcium concentration. This phenomenon is called pseudohypercalcemia (or factitious hypercalcemia).
In contrast to changes in the serum albumin concentration, alterations in pH affect the ionized but not the total calcium concentration [6]. Acidosis decreases the amount of calcium bound to albumin, whereas alkalosis increases the bound fraction of calcium; therefore total and corrected calcium are often inaccurate, particularly in critically ill patients [32]. The diagnosis and differential diagnosis of hypercalcemic conditions can therefore be improved by the availability of ionized calcium. The diagnostic workup of hypercalcemia also includes measurements of serum PTH, phosphorus, alkaline phosphatase, 25-OH-cholecalciferol and urinary calcium excretion. In some laboratories PTHrP is also measured.
As reported above, pHPT and malignancies are the most common causes of hypercalcemia, accounting for 80–90% of cases [1]. Hence, the diagnostic approach to hypercalcemia typically involves distinguishing between these two conditions. Usually patients with pHPT have elevated/normal PTH levels, while patients with cancer-associated hypercalcemia have suppressed PTH, as a secondary effect of elevated serum calcium concentrations. It is unusual for a patient with malignancy to have increased levels of PTH. Should this occur, a concomitant hyperparathyroidism or the secretion of PTH by malignancy itself could be suspected, although the latter is an uncommon event [6].
Therefore, when an abnormal total calcium result is obtained and confirmed by ionized calcium measurement, the PTH assay plays a crucial role in order to differentiate PTH-mediated from non-PTH-mediated hypercalcemia [33]. According to the results of the initial evaluation, a diagnostic flow-chart may develop, to differentiate the various conditions responsible for hypercalcemia, as depicted in Fig. 5.1.
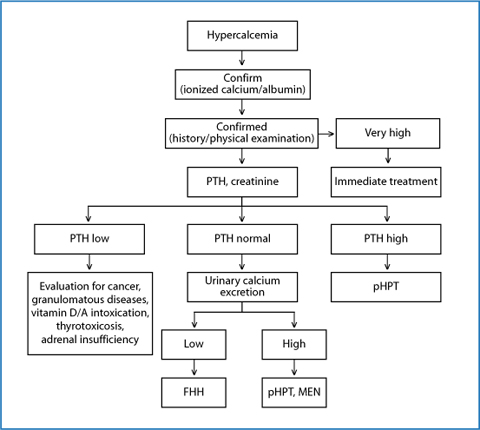
Fig. 5.1
Diagnostic workup of hypercalcemia. FHH, familial hypocalciuric hypercalcemia; MEN, multiple endocrine neoplasia; pHPT, primary hyperparathyroidism; PTH, parathyroid hormone
5.4 Etiology and Pathogenesis of Primary Hyperparathyroidism
Primary hyperparathyroidism may result from adenoma (single gland disease, accounting for 80–85% of cases; adenomas involving two or more glands are definitely less common), hyperplasia (multiglandular disease, accounting for 10–15% of cases), or carcinoma (less than 1% of cases, which typically involves a single gland) [1].
5.4.1 Parathyroid Adenomas
Parathyroid adenomas are benign neoplasms composed of chief cells, oncocytic cells, or transitional oncocytic cells (more of these cell types are frequently recognizable in the single lesion). They usually grow from normally situated glands, but ectopic localization, both in the neck and mediastinum, is not unusual (This aspect is extensively dealt with by Gasparri – see below) [34].
The average weight of adenomas is approximately 1 g, but many neoplasms are less than 0.5 g and microadenomas weighing less than 0.1 g are also well documented.
Larger adenomas are generally associated with higher levels of calcium and PTH and are more likely to be symptomatic.
Microadenomas are typically non-encapsulated, whereas larger adenomas often have a fibrous capsule. The cells of adenomas are arranged in cords, nests, sheets, and follicles and frequently are arranged around blood vessels. The nuclei are generally rounded with dense chromatin; they are usually larger than those of the normal parathyroid cells. Scattered pleomorphic and hyperchromatic nuclei, as well as multinucleate cells, are relatively common. Proliferative index, as demonstrated by Ki-67 assessment, is generally low. Immunohistochemistry of tumor cells shows positivity for cytokeratins, PTH, and chromogranin A.
Several adenoma variants have been described: oncocytic adenomas (composed of at least 90% of cells with abundant granular eosinophilic cytoplasm), lipoadenomas (hamartomas, composed of mature fat cells with foci of myxoid change, areas of fibrosis, and lymphocytic infiltration), water-clear cell adenomas (exceptionally rare). Adenomas may occasionally have a follicular architecture, similar to thyroid neoplasms [35].
Recent studies indicate that adenomas represent clonal expansions. One of the earliest described molecular abnormalities leads to overexpression of cyclin D1 protein [36].
5.4.2 Parathyroid Hyperplasia and Heritable Hyperparathyroidism Syndromes
Distinguishing asymmetric hyperplasia from adenoma may sometimes be extremely difficult. Hyperplasia is sporadic in approximately 75% of cases, whereas 25% of cases are heritable [37]. It represents the result of the absolute increase in parenchymal cell mass, with cells distributed in diffuse and/or nodular patterns; stromal fat cells are reduced. The weight of the gland ranges from 150 mg up to 10 g.
Familial HPT is a group of heterogeneous disorders including multiple endocrine neoplasia type 1 (MEN1), MEN2, FHH, neonatal severe hyper-parathyroidism, hyperparathyroidism–jaw tumor (HPT-JT) syndrome, familial isolated hyperparathyroidism, and autosomal dominant mild hyperparathy-roidism or familial hypercalcemia with hypercalciuria [38].
MEN1 is inherited as an autosomal dominant trait characterized by the development of multiglandular parathyroid tumors (90%), gastroenteropancreatic neuroendocrine tumors (60%), and pituitary adenomas (30%).
MEN2 occurs as an autosomal dominant trait characterized by the development of C-cell hyperplasia and medullary thyroid carcinoma (100%), adrenal pheochromocytomas (30%), and parathyroid hyperplasia (20–30%). A detailed description of these syndromes may be found in Chapter 14 [39].
Neonatal severe hyperparathyroidism represents the homozygous form of FHH. The disorder may be manifested at birth or within the first 6 months of life. Affected patients must be treated early with resection of the hyperplastic glands [9].
The HPT-JT syndrome is a rare autosomal dominant disorder due to mutations of the HRPT2 gene, which is characterized by hyperparathyroidism (multiple adenoma or parathyroid carcinoma in 10–15%) and fibro-osseous lesions of the mandible or maxilla. Renal lesions including cysts, hamartomas, renal cell carcinoma, and Wilms tumor can also be present [40].
Familial isolated hyperparathyroidism is characterized by the presence of benign multiglandular parathyroid disease, without other endocrine tumors. It has been associated with mutations in the MEN1 and CaSR genes and rarely with the HRPT2 gene.
5.4.3 Parathyroid Carcinoma and Atypical Adenomas
Parathyroid carcinoma is an uncommon tumor with the evidence of invasive growth (peritumoral vascular invasion, perineural invasion, invasion of adjacent soft tissues or thyroid). Most carcinomas have a solid growth pattern and mild to moderate nuclear pleomorphism, but some tumors may be indistinguishable from adenomas. The proliferative fraction of carcinomas is higher than that of adenomas.
Atypical parathyroid adenomas show some of the features of parathyroid carcinomas without frank evidence of invasive growth (peritumoral vascular invasion, perineural invasion, invasion of adjacent soft tissues or thyroid). They are characterized by intratumoral banding fibrosis, mitotic activity, trabecular growth, adherence of tumor to peritumoral soft tissues or thyroid gland, and presence of tumor cells within the surrounding capsule. Patients generally present calcium levels intermediate between those seen in patients with carcinomas and with adenomas. The clinical course is often benign [1].
5.5 Treatment of Hypercalcemia
The management of patients with hypercalcemia is dependent on the serum calcium levels, the rate of its increase, the signs and symptoms and the underlying disorder accounting for hypercalcemia.
For clinical purposes, hypercalcemia is generally considered mild when the serum calcium level is 10.5–11.9 mg/dL (2.6–2.9 mmol/L), moderate when calcium is 12.0 to 13.9 mg/dL (3.0 to 3.4 mmol/L) and severe when calcium is 14 mg/dL (3,5 mmol/L) or higher [6].
5.5.1 Treatment of Acute Hypercalcemia (Moderate to Severe Hypercalcemia)
If total calcium serum concentration is greater than 14 mg/dL, immediate treatment is warranted, regardless of symptoms, since this could be a life-threatening condition [41]. When hypercalcemia is moderate, the clinical picture is a guide to the therapy.
Management of acute hypercalcemia is similar in all patients and is independent of the cause. The therapy has four main goals: 1) to correct dehydration, 2) to enhance the renal excretion of calcium, 3) to inhibit accelerated bone resorption and 4) to treat the underlying disorder [30, 31, 42, 43].
5.5.1.1 Correction of Dehydration
Correction of dehydration is achieved by intravenous administration of isotonic saline (0.9%), 2–4 litres per day, with a urine output goal >2 litres per day. When depleted intravascular volume is restored, serum calcium concentrations decline to about 1.6–2.4 mg/dL (0.4–0.6 mmol/L). The increased filtration of calcium, by increasing glomerular filtration rate, decreases proximal tubular reabsorption and promotes calcium excretion. Hydration alone rarely normalizes serum calcium levels. Adverse effects are fluid overload and exacerbation of heart failure in patients with compromised cardiac or renal function.
5.5.1.2 Enhancement of Renal Excretion of Calcium
In addition to hydration with isotonic saline, therapy with loop diuretics may be indicated. Diuretic therapy should be considered after the correction of hypovolemia. Furosemide enhances calciuric effects of volume expansion, by inhibiting calcium reabsorption in Henle’s loop. Furosemide is administered at doses of 40–80 mg up to 500 mg/day. Adverse effects are dehydration and electrolyte abnormalities such as hypokalemia. An intensive therapy with furosemide requires frequent control of electrolytes and water excretion and is not necessary in most patients. Use of lower doses of furosemide (10–20 mg every 6 to 12 hours) are indicated to prevent fluid overload in older patients or in patients with compromised cardiac function.
Thiazide diuretics are contraindicated in severe hypercalcemia, since they may exacerbate hypercalcemia by enhancing calcium reabsorption in the distal nephron.
5.5.1.3 Inhibition of Accelerated Bone Resorption
Intravenous administration of bisphosphonates is highly effective in hypercalcemia induced by malignancies: pamidronate, zolendronate and ibandronate are the compounds of choice, due to their high potency and long-lasting effect. These agents lower serum calcium levels by decreasing osteoclastic bone resorption by two mechanisms: in the extracellular space, they bind to calcium phosphate and stabilize bone matrix; in the intracellular space they inhibit osteoclasts activity. Furthermore, they impair cellular ATP-dependent metabolic pathways, disrupt the osteoclast cytoskeleton and induce apoptosis.
Pamidronate (60–90 mg diluted in 200 mL of saline or 5% dextrose) is administered i.v. over 2 hours. Calcium levels start to fall within 2 days and the nadir is reached around the 6th day; the effect lasts 28–30 days. In patients with creatinine clearance <30 mL/min, infusion time should be slower (4–6 hours) and the dose should be adjusted for impaired renal function, since bisphosphonates are secreted unchanged by the kidneys through glomerular filtration.
Zoledronate (4 mg in 50–100 mL of saline or 5% dextrose) is infused i.v. over 15 minutes. A higher dose (8 mg) could be used in the case of relapse. Zoledronate is not recommended in patients with creatinine clearance <30 mL/min.
Ibandronate is administered i.v. at the dose of 3 mg. It is not recommended in patients with creatinine clearance <30 mL/min.
Adverse effects of bisphosphonates include renal failure and transient flulike syndrome with aches, chills and fever.
Calcitonin, a peptide secreted by the parafollicular C cells of the thyroid, inhibits bone resorption and also increases renal excretion of calcium. Salmon calcitonin is administered subcutaneously at the dose of 4 IU/kg up to 8 IU/kg every 12 hours. The fall in calcium concentrations (up to 1 mg/dL) is rather fast, reaching the nadir in 12–24 hours. Unfortunately, the effect of calcitonin is transient, due to the rapid development of tachyphylaxis within 48 hours, therefore it is only useful for a few days. Since the bisphosphonates take several days to become effective, a combination of isotonic saline, calcitonin and bisphosphonates may be helpful when rapid calcium lowering is warranted [30, 31].
Related posts:
 Management of Primary Hyperparathyroidism
Management of Primary Hyperparathyroidism
 Clinical Manifestations of Primary Hyperparathyroidism
Clinical Manifestations of Primary Hyperparathyroidism
 Surgical Anatomy of the Parathyroid Glands
Patient Information for Surgery in Primary and Secondary Hyperparathyroidism
Surgical Anatomy of the Parathyroid Glands
Patient Information for Surgery in Primary and Secondary Hyperparathyroidism
 Genetic Syndromes Associated with Primary Hyperparathyroidism
Genetic Syndromes Associated with Primary Hyperparathyroidism
 Preoperative Localization for Parathyroid Surgery in Primary and Secondary Hyperparathyroidism
Preoperative Localization for Parathyroid Surgery in Primary and Secondary Hyperparathyroidism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


