Chapter Outline
WHAT IS “CONSENT” IN PEDIATRICS?
ASSENT IN CHILDHOOD CANCER TRIALS
SPECIAL CONSIDERATIONS FOR CANCER TRIALS USING ALLOGENEIC HEMATOPOIETIC STEM CELL TRANSPLANTATION
MODELS AND TOOLS FOR ETHICAL DECISION MAKING IN PEDIATRIC CLINICAL TRIALS
FURTHER CHALLENGES IN PEDIATRIC CLINICAL ONCOLOGY TRIALS
Ethical decision making is a fundamental goal common to both clinical medicine and clinical research trials of all types. Broad attempts have long been made to thoroughly explicate the elements of ethical decision making that are common to both settings. However this goal has been pursued with increased vigor since the exposure of human moral abuses in the United States Public Health Service–sponsored Tuskegee Syphilis Study and by the Nuremberg trials and President Clinton’s Advisory Committee on Human Radiation Experiments. The practical yield has been the production of highly influential documents such as the Belmont Report, the Declaration of Helsinki and its several versions, guidelines produced for the International Arena by the Council for International Organizations of Medical Sciences, and most recently, the adoption of the Universal Declaration of Bioethics and Human Rights in October 2005 by the United Nations Educational, Scientific, and Cultural Organization. Common to these efforts are principles such as respect for persons, beneficence, and justice. Table 74-1 lists selected guidelines for research ethics. But how do general principles translate into practical action that is ethically sound for the practitioner faced with a particular clinical situation, for the clinical research physician considering whether to offer a phase I trial to a patient, and for the investigator whose nontherapeutic trials pose some risks?
| Guideline | Source | Year and Revisions |
|---|---|---|
| Nuremberg Code | Fundamental; Nuremberg Military Tribunal decision in United States v Brandt | 1947 |
| Declaration of Helsinki | World Medical Association | 1964, 1975, 1983, 1989, 1996 |
| Belmont Report | National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research | 1979 |
| International Ethical Guidelines for Biomedical Research Involving Human Subjects | Council for International Organizations of Medical Sciences in collaboration with World Health Organization | Proposed in 1982; revised, 1993 |
| 45 CFR 46, Common Rule | Other: U.S. DHHS and other federal agencies | DHHS guidelines in 1981; Common Rule, 1991 |
| Guidelines for Good Clinical Practice Trials on Pharmaceutical Products | World Health Organization | 1995 |
| Good Clinical Practice: Consolidated Guidance | International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use | 1996 |
| Convention of Human Rights and Biomedicine | Council of Europe | 1997 |
| Guidelines and Recommendations for European Ethics Committees | European Forum for Good Clinical Practice | 1997 |
| Medical Research Council Guidelines for Good Clinical Practice in Clinical Trials | Medical Research Council, United Kingdom | 1998 |
| Guidelines for the Conduct of Health Research Involving Human Subjects in Uganda | Uganda National Council for Science and Technology | 1998 |
| Ethical Conduct for Research Involving Humans | Tri-Council Working Group, Canada | 1998 |
| National Statement on Ethical Conduct in Research Involving Humans | National Health and Medical Research Council, Australia | 1999 |
The issues become considerably more complex when the patient or research participant is a child and the decision maker is not the person facing the possibility of risk or benefit. This issue poses special challenges in both pediatric clinical medicine and pediatric clinical research. The challenges are made more acute by the fact that they are not static. The considerations change as a child grows in age and experience and as we move from standard-of-care therapy to clinical trials, which, as recently argued, involve the standard of care for future children, the results of which have been a remarkable success story. Indeed it may be that survival percentages for adolescents and young adults with “pediatric” cancers have lagged behind those of younger patients in part because of their lower rates of participation in clinical trials. Further the issues do not remain static across all types of clinical trials. A child enrolled in a phase III clinical trial for newly diagnosed, standard-risk leukemia faces a situation very different from that of a child who has had multiple relapses of leukemia and whose family must choose between a phase I trial and a Make-A-Wish trip to the Grand Canyon.
This chapter begins with a brief historic review of the unique issues in pediatric medicine and pediatric clinical trials. It then moves to issues of parental permission and assent—issues that draw on, but are not fully informed by, the many studies of informed consent in adults. The differences in language and approach required as patients progress from phase III trials to phase I trials and palliative care are examined. After these more general issues, we will consider more specific questions, such as those raised by autopsy, stem cell transplantation, advances in technology, conflict of interest, insurance, ethics consultation, and international bioethics.
Historic Perspective
For hundreds of years, medical research has involved children as subjects. Edward Jenner first used an experimental smallpox vaccine on his own 1-year-old son at the end of the eighteenth century. At the end of the nineteenth century, a 9-year-old child was the first human recipient of Louis Pasteur’s rabies vaccine. However it was not until the first half of the twentieth century that regulations governing such experimental therapy began to be established and the importance of consent and parental permission became clearer.
The informed consent of human research subjects was first officially advocated in the Nuremberg Code, a code of ethics that grew out of the trial of German physicians who conducted egregious human experiments in Nazi concentration camps during World War II. However because this important code emphasized the absolute requirement of informed consent, it implicitly excluded children—who cannot give informed consent—from participating in research on human subjects.
The issue was more explicitly addressed in 1964, when the World Medical Association adopted a set of research ethics principles now known as the Declaration of Helsinki. This set of principles established the priority of the human subjects’ interests over those of science and society, and it sanctioned the participation of children in research if permission was given by the child’s responsible guardian.
Interestingly experiments were performed on children after the publication of the Nuremberg Code, which implicitly excluded children from research, and before the Declaration of Helsinki, which explicitly allowed such research under specific conditions. One of the most important of these experiments was a series of radiation exposure experiments. Among participating institutions was the Fernald School in Framingham, Massachusetts and the Massachusetts Institute of Technology with funding from the Quaker Oats Company, in which children deemed mentally retarded were fed radioactive iron and calcium in their cereal. President Clinton’s Advisory Committee on Human Radiation Experiments revealed the ethical consequences of inadequate parental permission in these experiments.
Another important set of experiments on children at that time occurred at the Willowbrook institution (located on New York’s Staten Island) and was designed to follow the natural history of hepatitis in children deemed mentally retarded. Newly arrived children were infected with hepatitis virus by housing them with others already known to be infected. Participating children were kept in a special unit with better conditions and nutrition, and children whose parents agreed to participation were admitted more rapidly. The lead investigator did require a thorough consent process that included a 2-week waiting period for full deliberation. However because Willowbrook was so crowded, critics subsequently argued that the expedited admission and special treatment amounted to coercion. These two cases underscored the need for ethical review and oversight of human subjects research involving children, even when parental permission is obtained.
The United States Congress became increasingly concerned about research ethics during the early 1970s, in part because of the syphilis study involving poor black men conducted by the U.S. Public Health Service at the Tuskegee Institute. As a result the National Commission for the Protection of Research Subjects of Biomedical and Behavioral Research was formed, and it published the Belmont Report in 1979. This report embraced three principles that are now familiar and accepted as crucial for research involving human subjects: respect for persons, beneficence, and justice. The principle of beneficence acknowledges the Hippocratic maxim “do no harm” and extends it to include maximizing the possible benefits and minimizing the possible harms to research subjects (minimizing harms is often formulated separately as a fourth principle, nonmaleficence). The principle of beneficence is operative in any discussion weighing the risks and benefits of research on human subjects, especially when the subject is a minor. The principle of justice concerns the right and fair distribution of the benefits of research, as well as the burdens, an issue that has a particular urgency when clinical treatment and research are considered globally. The concept of respect for persons includes two principles: that individuals be treated as autonomous agents and that those with less autonomy are entitled to protection. The latter principle is especially relevant in pediatrics and is somewhat fluid in its definition because of the growing autonomy of pediatric patients as they approach adulthood.
Given the past abuse of research subjects who were harmed in studies that they did not understand and for which they had not given meaningful permission, it is understandable that “informed consent” has become the dominant concept in discussion about ethical research on human subjects. However recent arguments have been put forth that informed consent is not always necessary for ethical clinical research, nor is it sufficient to qualify research as ethical. A recent review of major codes and declarations relating to research with human subjects led to the proposal that there be seven requirements for ethical clinical research. The research must lead to enhancement of health or knowledge. It must be methodologically rigorous. Selection of study sites and of individual subjects should be determined by scientific objectives and the potential for and distribution of risks and benefits. Given standard clinical practice and the research protocol, risks should be minimized, the potential for benefits should be enhanced, and risks must be outweighed by potential benefits to individuals and knowledge for society. Individuals should be informed about the research and provide voluntary consent. Research subjects should have their privacy protected, have the opportunity to withdraw, and have their well-being monitored. These requirements are thorough and persuasive, but they still must be adapted to the conditions under which the research is conducted. Factors of health, economy, culture, religion, and technology will affect how these requirements are translated into concrete, ethical action. For this reason, the specific context in which the research is conducted will affect ethical deliberation.
Ethical decision making in therapeutic and research efforts for children comprises all of the previously discussed considerations, plus the profoundly salient fact that the recipient of the risk or benefit is not the person who makes the decisions. The minor may not be capable of assimilating sufficient information to meaningfully participate in an “informed choice.” One need not enter the arena of international bioethics to find challenges to the role of “autonomy” in ethical decision making: such challenges are the daily fare in pediatric medicine and research, because children’s “autonomy” is an issue that changes from day to day.
What is “Consent” in Pediatrics?
The Nuremberg Code provides one starting point for understanding the meaning and importance of informed consent. The statement in this code that “the voluntary consent of human subjects is absolutely essential” has been interpreted as legal capacity, a power of free choice based on knowledge and comprehension. Consent, which was once seen as a single event, has come to be understood to be more of a process. It is not surprising, with the development of guidelines and goals for consent, that the process has become more intentional and more highly scrutinized.
The dominant theoretical framework for morally valid informed consent requires that four criteria be met: disclosure, understanding, voluntariness, and competence. Briefly, the core set of information that must be disclosed includes (1) facts (such as risks, benefits and alternatives) that patients (subjects) and providers believe are relevant to the decision, (2) the recommendation of the professional, and (3) the purpose, nature, and limitations of consent. Understanding goes beyond disclosure because, although the elements disclosed can be objectively stated, true understanding involves many variables and is more difficult to assess. Establishing and documenting understanding remains a great challenge, because information that has been disclosed but not understood contributes little to the ideal paradigm of informed consent. Voluntariness —another complex notion susceptible to interpretation—means at least that a decision is made without constraints of coercion or manipulation. Finally, competence , which is conventionally understood as the ability to perform a task, has also become a complex concept the definitions of which derive from law, psychiatry, and philosophy. At base a person’s competence (or perhaps more accurately expressed as cognitive capacity) to make a particular decision relates to the person’s ability to understand and think rationally about available choices, to weigh the benefits and burdens and risks within the context of his or her life and values, and then to use that understanding and logic to make a rational decision.
In the context of complex pediatric medical therapy or research, the process of consent is influenced by the fact that parents are making high-stakes decisions on behalf of their own child. The Children’s Cancer Group (CCG) conducted studies that showed that a sense of pressure is perceived by parents when making the consent decision for intervention in the context of childhood cancer. However a more recent study indicated that despite significant dissatisfaction with the consent process among CCG clinician-investigators, most parents were satisfied with it. The same study indicated that the parents’ satisfaction did not constitute adequate informed consent.
Even among clinicians assessment of the process of informed consent varies. The experience of the clinician is relevant in this assessment. Simon et al. found that clinicians with 10 or fewer years of experience were more likely to say that the most important goal of informed consent is to explain the disease and treatment and were more likely to suggest to parents that other children might benefit from the research. The same study found that in the end, when reports from clinicians and parents are compared, clinicians are dissatisfied with aspects of consent that parents seem far more satisfied with. For example, clinicians expressed concern about information overload and about the fact that the consent discussion often occurs while the parents are still in a state of shock about the diagnosis.
Further insights into this difference have been gained from a number of studies addressing the perspectives of parents of children with cancer. One such study used three focus groups of parents of children with cancer to retrospectively examine their perceptions of the informed consent process. High levels of stress were consistently reported, which were attributed to efforts to cope with multiple demands, including assimilating their child’s diagnosis, nurturing and supporting the child, understanding the information given to them about the diagnosis and treatment, getting to know an entirely new group of people involved in the child’s care, and participating in the child’s treatment. Another important point was that the parents did not consistently distinguish research from their child’s medical treatment. This finding underscores the importance of clearly explaining that research is optional during the process of “informing.” If no distinction is made between research and treatment, the goal of informing has not been met. Physician-investigators often experience tension between their role as the patient’s physician and their role as a researcher offering participation in a clinical trial designed to contribute to generalizable knowledge. This tension suggests that someone besides the physician-investigator should conduct the consent process. The study also suggested that the nature of clinical research, as well as the difference between research and proven current therapy, might be difficult for parents to grasp.
These conclusions were supported by a second study in which parents of children with newly diagnosed cancer were interviewed. All of the participants recalled the diagnosis, and most (80%) recalled survival statistics. However only slightly more than half knew that the treatment protocol involved research and understood the concept of randomization. This finding is striking when coupled with the fact that three fourths of the parents thought that alternatives to enrollment in a randomized protocol had been insufficiently discussed. Because randomization is such an important tool for answering certain clinical questions, this finding suggests that greater emphasis be placed on explaining unfamiliar concepts during the process of “informing.” Again, in this study, the majority of participants were satisfied with the consent process.
A study by Kodish and colleagues confirmed the difficulty of effectively conveying the meaning of randomization during the consent process in pediatric leukemia trials. In this multisite study, informed consent conferences were observed and audiotaped and were then compared with information acquired in interviews with parents shortly after the conference. The investigators found that although randomization was explained in 83% of cases, 50% of the parents did not understand it. Further, parents who did not understand randomization were more likely to consent to the randomized study than did those who did understand it, although this difference was not statistically significant ( P = .07). These findings led to several recommendations for improving understanding, including a clear explanation of the differences between the randomized trial and off-study therapy, assessment of parental understanding of randomization, and further explanation when the idea was not yet grasped.
Other efforts have been made to improve the process of informed consent in pediatrics, especially in the case of complex clinical trials. For example, a study by the Children’s Oncology Group (COG) assessed the possibility of staged consent; investigators had the option of obtaining consent over a 28-day period using a staged approach. This option allowed parents and patients more time to discuss and absorb facts about the disease, the purpose of the trial, the design of the study, and the potential risks. Several measures in this study suggested benefit from the staged approach. There was greater understanding of treatment choices and of the distinction between a randomized controlled trial and standard therapy with the staged approach (80% understanding) than in the other studies (62.5%, P = .05).
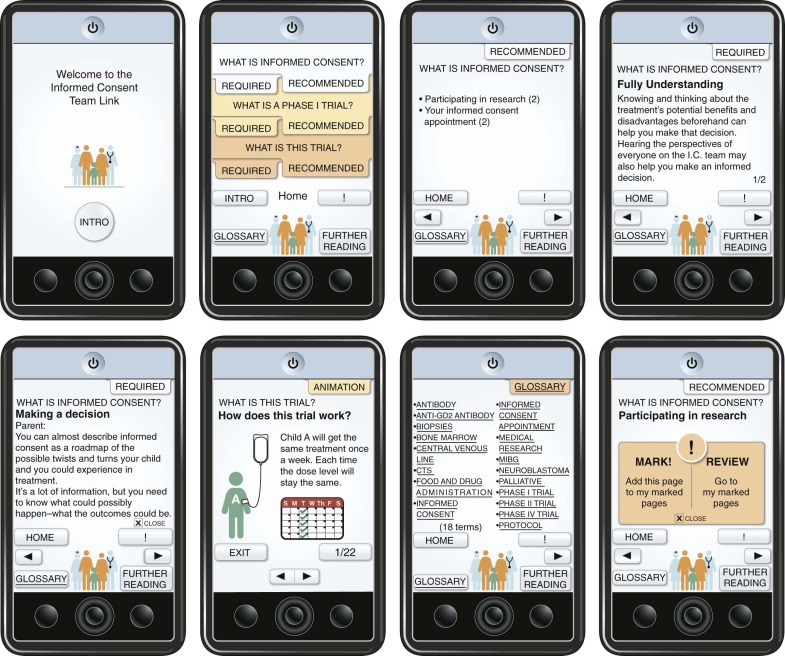
Contemporary medicine will require the introduction of new methods of informing patients and parents to meet the ethical imperative of informed consent in pediatric trials. Informed consent documents for cancer clinical trials are sometimes long and difficult to understand, with language that serves less to protect human subjects than to decrease institutional liability. Therefore some advances may take the form of technologic and educational methods to explain ever more complex medicine and research to people not formally trained in a medical discipline. One such example is a device known as the Informed Consent Team Link ( Fig. 74-1 ). This device was designed to help families understand phase I trials, which present daunting challenges to fully informed consent. The design was based on information from focus groups of parents, physicians, nonphysician health care workers, and teenaged patients. Among the elements considered important were (1) the need for a “big picture” overview that would create a context for details, (2) delivery of information in small chunks, (3) use of multiple modes of information delivery, and (4) prioritization of information. Such basic observations provided a basis for the design of a device that uses interactive technology, voice, animation, and health education principles to deliver information about phase I trials. Because of children’s apparently natural affinity for interactive electronic devices, this approach may be helpful in addressing assent as well.
Concepts about the nature of consent in pediatrics are evolving with changes in contemporary medicine. For example some persons argue that informed consent may be too restrictive a concept and that “valid consent” should be substituted. The three aspects of valid consent are personal competence (Does the patient have the capacity to make the decision?), procedural competence (Is the consent given correctly?), and material competence (Is the procedure consented to appropriate for valid consent?). The concept of valid consent has been explored in the pediatric context by the Society for Industrial and Organizational Psychology Working Committee on Psychosocial Issues in Pediatric Oncology. The value of this notion, they suggest, is that it emphasizes the patients’ or parents’ understanding of what is being consented to and recognizes that there are both rational and nonrational aspects to the decision-making process that must be understood. The underlying concern is that informed consent has come to mean legally signed documentation rather than real understanding, insofar as that is possible. Because parents come from different backgrounds and because children of different ages differ in their ability to grasp complex concepts, the level of understanding that is attainable may vary from situation to situation. The notion of valid consent acknowledges this fact and counters the idea that a signed document is equivalent to informed consent.
The four elements of informed consent previously mentioned—disclosure, understanding, voluntariness, and competence—are useful for informed consent in adults, but informed consent in pediatrics is complicated by the fact that three parties are involved (parent, child, and clinician/investigator) and that the research subject is the child. In pediatrics, it is not autonomy (the basis of the four components of consent) that takes precedence but rather the best interest of the child. The best-interests notion is clearer in a purely clinical setting than in a research setting, in which interventions are designed to contribute to general knowledge. Despite the difficulty of approximating truly informed consent in the setting of pediatrics, and especially pediatric research, the obligation to advance pediatric medicine lends urgency to continued efforts to offer the closest possible approximation to informed consent.
One important aspect of the difference between informed consent in adult and pediatric medicine and research is the fact that in pediatrics, informed consent is better thought of as a combination of parental permission and (as appropriate) the more complex concept of the assent of the child.
Assent in Childhood Cancer Trials
The ethics of assent is one of the most difficult issues faced in pediatric clinical trials. Many adolescents, and even some younger children, possess the elements of competence or health care decision-making capacity (setting aside for the moment the legal definitions, which hinge on age rather than capacity). This is especially true, perhaps, of children who are exposed to long-term clinical trials and to the environment of a children’s hospital for long periods. Certainly younger children are not developmentally capable of comprehending complex protocols, but they do have some level of understanding that increases with age and experience. How should assent be understood and when should it be required? But first, what do we actually mean by “assent” and how can it be distinguished from consent?
Consent refers to an active affirmative agreement to something by an autonomous agent (i.e., a person). It is capable of being given by anyone who meets the minimal criteria of being in the state of mind and body to be an autonomous agent; generally, most adults would qualify as long as they were not placed in a situation in which their decisions might be suspected to be the result of some form of coercion. The ability to give consent is also context and subject dependent in the sense that most people are generally not competent in all spheres of their lives: for instance, many persons who are perfectly capable to make health care decisions or even agree to participate in research may not be competent to make (informed) decisions about automobile repair. For the purposes of this discussion, the relevant area in which cognitive capacity is important is in the domain of health care (and associated human subjects research). In the United States the legal age of consent is 18 years for most realms (nearly all jurisdictions, however, permit older adolescents who have not yet reached the age of 18 years to make some types of health care decisions [i.e., give consent], usually in regard to reproductive medical issues). Assent is a more complex concept; in general it describes a less active form of agreement to a proposed procedure or action, rather than the more active and involved consent. It is used in an attempt to engage children and adolescents in the process of consent that must be given by parents or other authorized surrogate decision makers. However what may qualify as acceptable assent (a grudging or sullen nod of the head or a more energetic and participatory form of positive interaction) remains an area of reasonable concern.
Far less is known about assent than about consent. Much remains to be learned about the practices of institutional review boards (IRBs), the perceptions of parents, clinicians, and children about consent, and the methods that might be most effective in the process of assent. Several studies have looked into these issues and documented the need for further work to address variability in IRB practices and the implementation of assent. One study found that half of IRBs have a method they require investigators to follow when determining which children are capable of assent, whereas half have no such method, and the majority of IRBs rely on investigators’ judgment. IRBs need guidance on the implementation of requirements regarding assent. Authors of a second study compared standards for assent, as well as consent forms approved by 55 local IRBs, when reviewing three standardized multicenter research protocols. Standards varied widely; 35 had separate forms and simplified language for assent, and 31 specified lower age ranges for obtaining assent in three studies. For a hypertension study, the age at which assent was required ranged from 6 to 15 years;, for a pain study, the age range was 6 to 12 years; and for a respiratory failure study, the range was 7 to 12 years. It is not clear why some IRBs consider a child of 6 years to be capable of assent while others do not require assent until a child is 15 years old.
Do the regulations help us understand assent? Most research on children is governed by subpart D of 45 Code of Federal Regulations (CFR) 46, which provides additional protections for children (as vulnerable subjects) beyond those specified in subpart A of 45 CFR 46. Subpart D, added in 1983, outlines three categories of research that a local IRB can approve: research not involving greater than minimal risk to participants (45 CFR 46.404), research involving greater than minimal risk but with the prospect of direct benefit to individual participants (45 CFR 46.405), and research involving no greater than a minor increase over minimal risk (with no prospect of direct benefit) but likely to provide generalizable knowledge of the subject’s condition or disorder that is vital to understanding or ameliorating it (45 CFR 46.406; Table 74-2 ). Local IRBs cannot approve research that does not fall into one of these categories, and performance of the research is possible only if two conditions are met: (1) the local IRB finds that the research provides a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health and welfare of children, and (2) the protocol is approved by the Secretary of Health and Human Services, after soliciting the opinions of an expert panel and providing for a period of public comment (45 CFR 46.407). According to these regulations governing research on children, assent means “a child’s voluntary affirmative agreement to participate in research. Mere failure to object should not, absent affirmative agreement, be construed as assent” (45 CFR 46.402). If the IRB determines that the participants in certain categories of age and maturity are capable of providing assent, investigators must obtain it to proceed. When assent is required, a child’s refusal is generally binding.
| PROSPECT OF DIRECT BENEFIT | ||
|---|---|---|
| Level of Risk | No | Yes |
| Minimal risk | Approval by IRB permitted (46.404), conditioned on:
| |
| Greater than minimal risk | Approval by IRB permitted (46.406), conditioned on:
| Approval by IRB permitted (46.405), conditioned on:
|
| Not otherwise approvable, but presents the opportunity to understand, prevent, or alleviate a serious problem affecting the health or welfare of children | Approval requires U.S. Department of Health and Human Services approval after consultation with an expert panel and opportunity for public review and comments (46.407), conditioned on:
| |
Can an IRB waive the requirement for assent? Yes, but only if (1) the intervention or procedure offers the possibility of direct benefit that is important to the health or well-being of the child and this intervention or procedure is only available in the research context (45 CFR 46.408[a]) or (2) the IRB determines that children below a certain age in a certain situation or with a certain condition have such a limited capacity to participate in the decision that they cannot reasonably be consulted.
The requirements are difficult to apply, especially in the context of clinical trials for childhood cancer. Most of the children are enrolled in research studies in which the therapy is prolonged and carried out in the context of profound physical, spiritual, and psychological stress. The Bioethics Committee of the COG convened an international multidisciplinary task force to address assent in 2003. This group identified a number of problems with the regulatory framework. In brief the regulations do not take adequate account of the dynamic moral and cognitive development of children and thus allow a child either to have no formal role in decision making or to have the power of veto. Nor do the guidelines make clear what constitutes meaningful assent, and so they leave IRBs and investigators with uncertainty about when they have or have not hit the mark (although other groups have provided guidance in this area). The regulations do not take into account the fact that some clinical research is more complex than other research, and thus some research may be more accessible to the understanding of children than other research. The regulations say that permission from the guardians and assent from the child are distinct decisions, and they do not take into account the manner in which parents and children make decisions together. Finally, cultures in which autonomy is less emphasized are not accommodated by a regulation that potentially sets parental prerogatives against a child’s veto power.
Taking into account these factors, the COG Task Force offered three principles governing children’s participation in research decisions ( Box 74-1 ). A number of specific recommendations followed from these principles. The Task Force acknowledged that in some settings the principles offered will sometimes conflict. Therefore the recommendations emphasize the importance of establishing procedures for the resolution of conflict rather than attempting to define universal rules. In that way the absence of universal rules governing decisions in families with, perhaps, different cultural backgrounds and assumptions does not end conversation and negotiation. This approach is especially valuable in an increasingly pluralistic society and a global culture of information sharing. Much work remains to be done to understand the processes of communication and learning and the interactions that comprise the permission-assent process.
- 1.
Investigators should always respect children as persons. In particular, investigators together with parents should honor children’s developing autonomy in decisions about research.
- 2.
Investigators should respect parents’ roles in guiding their children’s moral development and assessing their best interests. For example, parents should have discretion to determine the degree to which children should be encouraged to participate in activities that benefit others.
- 3.
Policies regarding assent, as well as decisions of Institutional Review Boards with respect to particular protocols, should be sufficiently flexible to accommodate the wide range of medical, psychological, and contextual circumstances seen in pediatric oncology.
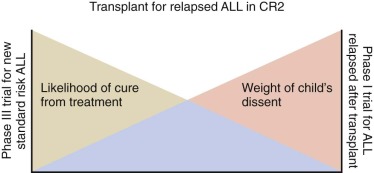
The flexibility of the approach using conflict resolution acknowledges that the obligation to obtain assent may change, depending on the time and situation, even for the same child. For example assent considerations may differ between a 9-year-old child with standard-risk leukemia who may obtain considerable benefit from enrollment in a phase III leukemia trial and a 9-year-old child who has leukemia that has relapsed multiple times and for whom a phase I trial is being considered. Similarly, a child at the beginning of a 2 and one half-year course of therapy for leukemia will be different in age, maturity, vocabulary, experience, and capacity to understand by the end of that course of therapy ( Fig. 74-2 ). Developmentally appropriate approaches to help the child understand the experience do not have the restrictions and limitations that accompany the binding aspects of assent.
For older adolescents assent should be approached in the same way as adult informed consent, even though parental permission may be required. Some adolescents may have the capacity for consent but may choose to have their parents make decisions about their participation in research. Several studies have recently assessed assent in adolescents. Our understanding of the differences between parents’ and adolescents’ perceptions of research is growing as we learn more about the assessment of risk (i.e., concern for physical safety) versus aversion (psychological discomfort). More than 3000 adolescents in the United States alone die of chronic illness and cancer each year. In such situations when adolescents meet the criteria for functional competence, the broad ethical consensus is that decisional authority should be granted to them regardless of their legal decisional status. However this is an area in which a wide divergence exists between what ethics may demand but the law allows. Most states and jurisdictions do not permit minors to be sole participants in medical decision making, including consenting to participate in research, unless they have been emancipated according to the laws of the state in which they reside. This restriction does not imply that adolescents should not fully participate in the decision-making process, and there certainly is no legal proscription against their doing so; it simply underlines the difference between good ethical clinical (and research) practice and the legal framework. It might perhaps be better stated that in these situations, if the adolescent is going to be a research subject, assent is necessary but not sufficient.
Pediatric Clinical Trials
The majority of children with cancer are enrolled in clinical trials. This systematic evaluation of interventions is largely responsible for the dramatic improvement in survival during the past 40 years. A number of important ethical concepts influence the design and execution of these trials. Permission and assent have already been discussed. Other important issues include the distinction between therapeutic and nontherapeutic research, the various concepts of minimal risk and clinical equipoise, and the considerations that distinguish phase I and II trials from phase III trials.
As previously described and in Table 74-2 , reports published by the National Commission for Subjects of Biomedical and Behavioral Research formed the basis of current regulations governing research with human subjects, including children. One of the most important distinctions made was between research that does, versus does not, offer the prospect of direct benefit to the child. Some elements of a clinical trial may offer the prospect of direct benefit (for example, chemotherapy for acute lymphoblastic leukemia), whereas other elements may not (e.g., biologic and pharmacokinetic studies designed to improve future trials). What are the general criteria used to determine whether the latter sorts of research are permissible?
Minimal Risk
The central ethical concept that guides discussion of pediatric research that has no prospect of direct benefit is “minimal risk.” The Belmont Report defines minimal risk as “the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical or psychological examination, of healthy children.” The question that immediately arises is which children’s lives should be the basis for deciding what is “normally encountered in daily life.” These risks can vary widely, not only from country to country but within subgroups in a single country. Even if the notion of “harms or discomforts that average, healthy, normal children may encounter” (as clarified by the National Bioethics Advisory Commission) can be roughly agreed upon as a standard, the question remains whether it is ever acceptable for a parent to give permission for any risk greater than minimal risk in the absence of direct benefit to the child.
Because of concern that the “minimal risk” standard would exclude some pediatric research that is important and appropriate, the National Commission allowed parental discretion in giving permission for research that exposes children to a “minor increase” over minimal risk, given the fact that parents routinely permit their children to participate in activities, such as contact sports, that present more than a minimal risk, and given the potential benefits of such research to others. Therefore a “minor increase” over minimal risk is permissible if the proposed research involves only procedures that such children might normally experience because of their medical condition and if the proposed research has the potential to yield vitally important knowledge for understanding or ameliorating the child’s particular condition.
The permissibility of a “minor increase” over minimal risk provides a conceptual way to avoid barriers to important pediatric research. However a difficult question remains: exactly what constitutes a “minor increase”? The term is used in the federal regulations, but it is not defined. Wendler and Emanuel proposed five possible standards for defining a “minor” increase consistently among IRBs. These standards include (1) a fixed percentage above daily risks, (2) the confidence intervals around the risks of daily life, (3) the risks an ill child might encounter in an examination, (4) the “scrupulous parent” standard, in which minor risks are sufficiently similar to the child’s past experiences in daily life, and (5) the “socially acceptable” risk standard.
Wendler and Emanuel argue that the fifth standard is the most reasonable. The “socially acceptable” risk standard takes as its starting point the fact that some children encounter greater risks in daily life than others. The socially acceptable risk standard allows risks that are greater than those faced every day by healthy children, but it limits the risks to those that are socially acceptable. The socially acceptable risks greater than those experienced by the average healthy child may be more closely defined through research. In any case this standard tethers the concept of “minor increase over minimal risk” to an evaluation that is nonarbitrary, that does not impede important beneficial research, and that does not justify greater risks for ill children simply because their daily lives are filled with more risk (due to their disease and its treatment). An alternative to the approach taken by Wendler and Emanuel has been suggested by Nelson and Ross. They argue that the “scrupulous parent” standard is ethically justifiable and that such an approach incorporates both minimal risk and a minor increase over minimal risk within a uniform standard that should be applicable to all children.
Phase I and II Trials in Pediatric Oncology
Phase I studies have the unique primary objective of assessing the maximum tolerated dose and estimating toxic effects in human subjects, in contrast to phase II and phase III studies, which explicitly assess the efficacy and relative safety of interventions. Phase I studies in children are important because the pharmacokinetics and toxic effects of drugs differ between children and adults. Therefore if drugs were used in the general pediatric population without phase I testing in children, the incidence of adverse effects would be unpredictable. Additionally some diseases are unique, or nearly unique, to the pediatric population, including a number of pediatric cancers. Prohibition of phase I studies in children would limit the ability of the research community to develop new drugs that target uniquely pediatric diseases.
Phase I studies are typically dose-escalation trials in which some children receive a dose unlikely to be of any benefit while others receive a dose that is toxic. Therefore phase I studies do not fit the category of minimal risk nor, many would argue, of a “minor increase” over minimal risk. Much of the controversy about phase I studies in children, therefore, centers on whether they offer sufficient direct benefit to justify the risks, both from a purely ethical point of view and from the regulatory point of view.
What is the likelihood of harms and benefits in phase I oncology trials? A recent review of abstracts and reports of the results of phase I cancer trials submitted to the American Society of Clinical Oncology (ASCO) from 1991 to 2002 found that the death rate from toxicity was 0.54%, with an overall objective response rate of 3.8%, and that risks decreased over time. A National Cancer Institute–sponsored study of adult phase I oncology trials run between 1991 and 2002 found an overall response rate of 10.6%. The response rates in “classic” single-agent phase I trials were lower (4.4%), whereas those that included at least one Food and Drug Administration (FDA)–approved anticancer agent had a response rate of 17.8%. In pediatric oncology phase I trials, the response rate (both partial and complete responses) has been reported to be between 4% and 9.6%. However the (causal) relationship or correlation between dose and response is rarely reported, and the small number of subjects at any dose level would preclude any sort of definitive conclusion from being drawn. The overall rate of death due to toxicity was 0.49%. That said, in any single phase I trial centered around a single drug, considerable variability in outcomes can exist among the research subjects recruited. Hence it is reasonable to conclude that few children entered in phase I trials die from study-drug–related toxicity and a very small percentage may derive some therapeutic benefit. Unfortunately no feasible mechanism exists to abstract what might be called the “pain and suffering” effects of trial participation or whether—as has been reported for adults enrolled in similar studies—there may be an avoidance of engaging in appropriate end-of-life discussions and planning. Finally, at least one study has suggested that parents may feel compelled by their child’s physician to enroll in phase I trials. These observations suggest the idea of “collusion” between doctors and patients to temper the discussion of bad news with the hope that may be imparted by treatment.
Can phase I trials be viewed as offering a prospect of direct benefit in the context of pediatric oncology? The pediatric medical and bioethics communities are divided on this question. The fact is that most people who enroll in phase I trials hope for personal benefit, although they know that the trial is designed to test toxicity. The concern exists that such a “therapeutic misconception” may encourage false hope for cure and lead parents to make decisions that do not take into account all of the elements that make up “the child’s best interest.” This phrase refers to the mistaken belief by clinical trial subjects (especially those participating in early-phase studies, such as phase I) that the principal purpose or goal of the trial is therapeutic, rather than data generation. Clearly the primary scientific goal is not therapeutic, and the fundamental purpose of these trials is to gather data on toxicity, physiologic tolerability, and dosage. However it is very difficult to communicate such bare facts to families. Much of the confusion is derived from the emotional tension of the situation, the hopes and fears of parents (and patients), the fact that treating physicians often are involved in discussing the clinical options available for terminally ill patients, and the mixed messages that are both transmitted and received by families during these conversations. The simple fact of the matter is that parents may not wish to hear “the truth” and their physicians may not wish to transmit it. Although many persons consider this problem to be almost insuperable, some attempts have been made to combat it with mixed success. Nevertheless it remains a significant complicating factor that contributes to the ethical complexity of performing pediatric phase I trials.
On the other hand because only patients for whom known therapies have failed are usually eligible for phase I trials, these trials may be viewed as offering a relative prospect of direct benefit, although the chance of controlling the disease is small. Such a view would bring phase I research under CFR 46.405 of the federal regulations, but it has been criticized because such a classification (in which risk is justified by potential benefit) fails to take into account the researcher’s focus on safety rather than efficacy. From the latter point of view, phase I research would be approvable only under CFR 46.407, in which the research is reviewed and approved by a panel appointed by the Secretary of the Department of Health and Human Services—a time-consuming effort that requires both national and local IRB review.
One critic of the “direct benefit” argument also questions the approval of phase I trials under CFR 46.407 because the required review process delays providing benefit to children and adds little to the meaningful protection of children. This author has suggested that a new category of research offering the potential for “secondary direct benefit” be added to the regulations. Such a category would require (1) that the risks be justified by the likelihood that the research will yield generalizable knowledge, (2) that the research be commensurate with the lived experience of those with the disease or condition, (3) that the research offer a potential secondary benefit that is otherwise not available (a potential therapeutic benefit even though the trial lacks therapeutic intent), and (4) that consent requirements be met. Work that addresses gaps in the understanding of phase I trials and the federal regulations governing them should take high priority in pediatric medical ethics research. A recent essay supports phase I trials in children using an interesting and somewhat novel argument. Morris suggests that most children who would be eligible for the types of phase I trials that may involve more than minimal risk (independent of how it may be defined) would of course be afflicted with the disease or condition for which the agent under study is under investigation (for future possible therapeutic use). Furthermore it is likely that the experience of this disease would have acquainted both the child and his or her parents with many of the tests and procedures used as part of the trial (such as phlebotomy). Because they would also have an inherent interest in the outcome of such studies (both for the present and the future), they (and local, institutional IRBs) should be extended some moral flexibility in interpreting what should be interpreted as “minimal risk.”
Phase II trials play an important role in all areas of cancer therapy. They provide the initial assessment of treatment efficacy and identify agents for further investigation in phase III studies. These studies, although experimental, are clearly therapeutic in intent and thus the therapeutic misconception is not as germane. Phase II studies are likely to increase in number as high-throughput screening of compounds identifies more candidate anticancer drugs and more molecularly targeted agents and biologic agents become available. This fact will raise many important ethical questions in pediatric clinical trials, because phase II studies specifically evaluate drug efficacy before benefit is clearly demonstrated. Therefore it is vital that families and patients be fully informed about alternatives.
We should mention the attraction for incorporating nonrandomized phase II trials into therapeutic phase III studies, the so-called “phase II window.” This approach was quite popular for some time because of the belief that previously untreated, “chemotherapy-naive” patients would constitute a population that would better represent the efficacy of a given agent compared with heavily pretreated patients whose response rates might be diminished both because of cross-resistance to other drugs and decreased performance status. This technique has come under significant ethical and statistical criticism, and in 2010, a consensus statement from the National Institute Clinical Trial Design Task Force was issued that called for the elimination of single-arm studies of this type and their replacement with better designed trials (although the Task Force did express the caveat that unique situations may exist in which nonrandomized, window designs might be appropriate). Many stand-alone phase II COG studies have had this design, and it is rarely used in phase III studies (only one as of this writing). Therefore although phase II windows and single-arm studies still exist, the future of this form of design is questionable.
Finally, a word should be said about the use of drugs that are still under investigation in an “off-label” capacity. FDA-approved agents have been demonstrated to be safe and effective by FDA standards and thus receive approval for one or more indications. Many of these approved agents that have not yet been shown to have similar efficacy or a justifiable safety profile in children are nonetheless used in a phase II–like practice, but without the protections of an IRB-approved clinical trial (including rigorous and supervised informed consent and an adverse event reporting framework). Data about the frequency of this practice in children have not yet been published, but Peppercorn and colleagues have reported this practice among adult oncologists, even when a clinical study incorporating the same drug(s) and for which the patient is eligible is available. Although it may be imagined that many studies use drugs that are only available via the trial, this situation is not actually the fact in adult oncology. In pediatrics this observation would be even more germane because it is rare for an agent to be available for children (especially for phase II and III trials) without first being tested and approved in adults. The widespread use of this practice is worrisome for several reasons: First, it tends to undermine the clinical trial enterprise if physicians are willing to prescribe (off-label) investigational drugs in the same clinical setting in which they are being investigated. Such use also can lead to the failure to notice uncommon toxicities that may be more easily detected within the stricture of a formal clinical trial. Second, it is argued that such use results in the conduct of research without the significant human subjects oversight. Third, this practice exposes children to more than minimal risk, but the (relative) safety of patients cannot be ensured as it would be in a clinical trial with formal tracking of adverse events. Finally, this practice represents a missed opportunity for more phase II trials to be conducted with sufficient statistical power to enable substantive conclusions about their significance to be drawn. Thus this practice exposes patients (subjects) to potential risk without even the compensatory benefit of contributing valuable scientific knowledge to the general community.
Phase III Trials and Clinical Equipoise
If the concept of minimal risk demarcates the upper limit of nontherapeutic research, the notion of clinical equipoise is central (although not unproblematic) to ethical thought about randomization in clinical research. Clinical equipoise requires that if patients are to be randomly assigned to one of two interventions in a clinical trial, real doubt must exist—in the medical community as a whole, if not in the mind of the investigator—about which of the two interventions is superior. This requirement is said to provide one answer to the possible conflict of interest that clinical investigators can experience in the dual role of researcher and physician.
At least two sources of confusion are presented by all forms of clinical trials, especially in pediatrics. First, sick children are first and foremost patients, which creates a special and unique fiduciary relationship between the patient and his or her doctor. This relationship also establishes a duty on the part of the doctor toward the patient that confers certain responsibilities to act in the patient’s best interest. This relationship should be distinguished from that of an investigator to a research subject, which is defined within the parameters of federal regulations (previously described). In pediatric hematology-oncology, physicians often occupy both roles simultaneously, which can present significant conflicts (although not necessarily straightforward conflicts of interest). For example if the treating physician is also in some way involved in the design or implementation of the clinical trial under discussion with a patient and his family, she (the doctor) may have a conflict. Enrolling in the trial may not necessarily be to the best advantage or well-being of the patient as defined by him (and his surrogates) and his physician, but participating in the trial as a subject could be beneficial to the investigator (the doctor). In these situations it may be advisable for the treating physician to avoid caring for patients with diagnoses that may make them eligible for the trial for which he or she is responsible. For physicians whose relationship to the study is peripheral, in a manner that could perhaps be described as administrative (as is often the case for many, if not most, treating physicians with COG clinical trials), the conflict is minimal. However in a number of institutions, participation in “therapeutic” (i.e., phase II and III) trials may be remunerative, if not to the doctor, to the local group that employs the physician, by way of financial reimbursements to (partially) cover the cost of performing clinical trials. Although these rewards are hardly profitable, they do raise the specter of a conflict and may need to be addressed. A commitment to transparency may be sufficient in these cases. Second—and we admit that this may be more of a theoretical concern—years of experience have demonstrated the enormous benefit bestowed by the cooperative group clinical trial enterprise in pediatric oncology. Of course this progress would only have been possible with the recruitment (by physicians) and participation (by patients/subjects) in many clinical trials during the past four decades. The ongoing dedication of families, patients, and pediatric oncologists to this endeavor has been crucial to its success. Hence it could be perceived that there is a not-so-subtle pressure to enroll as many (eligible) patients as possible in group clinical studies (both therapeutic and nontherapeutic); indeed, failure to enroll sufficient numbers may result in institutional sanctions, up to and including suspension from the Group and thus an inability to access study protocols and documents (see the COG website for details). However, the recognition that this cooperative endeavor has been both productive and protective of patient (and subject) welfare is well understood and accepted. Nevertheless, the conflicts and the potential for their intensification and complexity remains and actually could increase as more pharmaceutical industry clinical trials are incorporated into the cooperative clinical trial group apparatus.
Does the physician’s “therapeutic obligation” require the provision of the best possible care or simply of competent care? The concept of clinical equipoise has been challenged from a number of perspectives. Some critics argue that therapeutic medicine and clinical research are distinct types of activity that require distinct ethical frameworks and that the concept of “clinical equipoise” cannot provide a unifying ethical framework. Clinical equipoise has also been criticized as especially problematic in developing countries, where the lack of resources may not support trials that test new treatments against established therapies or placebo. The requirement for clinical equipoise could make it difficult to justify the search for interventions that, although they provide significant benefit for less money, are known not to be the most effective interventions.
Several other considerations are important in evaluating the concept of clinical equipoise. First, the conditions for establishing true equipoise are not objective, based as they are on unverified judgments that are open to bias. Second, if clinical equipoise is not always a reliable foundation for randomized studies, how do we resolve the conflicts that can be posed by clinical investigators’ dual roles? Finally, although adults may be able to negotiate the ambiguities of clinical equipoise in a clinical trial, children cannot do so. In light of the previous discussion regarding the best interest of the child (or the attempt to avoid harm to the child), no phase III randomized pediatric trial should be conducted unless genuine doubt exists about which of two or more alternatives is better.
Recently a new approach to bringing together research and clinical care has begun to emerge. The Institute of Medicine has called for the transformation of health systems into “learning health care systems” that are able to study and continuously improve their practices. Learning health care systems pursue a wide range of investigations, including those aimed at quality improvement and better clinical effectiveness. An active question is whether the data collection and monitoring central to learning health care systems should be construed as research, and if so, what type of ethical oversight it should have.
Finally, a great deal of discussion has ensued about the responsibility of investigators to communicate the results of clinical trials to patients and their families, irrespective of the outcome of the studies. Surveys have suggested that patients who have enrolled in trials (or who have been enrolled by their parents) retain an interest in the outcome. Thus there is good reason to believe that a continuation of the respect we pay to patients, and the diligence and responsibility owed to study participants/subjects, would warrant the formal reporting of the results of trials to them, even taking into consideration the time lag that is intrinsic to the completion of a large study, the maturing of the data, and the challenges associated with communicating complex information of this type. Furthermore, the variety of data that can be collected from the trial—including intricate genetic and other biologic marker data—will require careful consideration to decide what information would be most important to communicate. The issue of conveying genetic or genomic information has proven to be particularly problematic, and to date the challenge has resisted consensus solutions.
Related posts:
 The Neonatal Erythrocyte and Its Disorders
The Neonatal Erythrocyte and Its Disorders
 Diagnostic Approach to the Anemic Patient
Diagnostic Approach to the Anemic Patient
 Approach to the Child with a Suspected Bleeding Disorder
Approach to the Child with a Suspected Bleeding Disorder
 Disorders of the Red Cell Membrane
Disorders of the Red Cell Membrane
 Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
Pediatric Myeloid Leukemia, Myelodysplasia, and Myeloproliferative Disease
 Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Nonrhabdomyosarcoma Soft Tissue Sarcomas and Other Soft Tissue Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


