Fig. 16.1
Graphical display of the most common epigenetic changes in CpG islands of tumor suppressor genes and DNA repeats in normal and cancer cells.
16.3 Technical Approaches to Detect Epigenetic Changes
Techniques for the detection of epigenetic changes have dramatically evolved during the last 30 years [39–43]. The initial efforts in the 1970s were focused on the measurement of global DNA methylation content and the study of particular sequences by Southern blot analyses using methylation-sensitive restriction endonucleases. The limitations of the latter method (e.g., large amounts of high-quality DNA, sequence biases, and problems with incomplete digestions) made the study of specific sequences time consuming and not widely applicable. It was not until 1992, with the introduction of the sodium bisulfite conversion technique, that DNA methylation analyses made a revolutionary step forward [44]. Sodium bisulfite has the property of converting unmethylated cytosine into uracil, whereas methylated cytosine remains unmodified. The combination of this chemical modification with genomic sequencing and methylation-specific PCR (MSP) made the study of DNA methylation changes widely available, and a large number of studies, especially dealing with CpG island hypermethylation of tumor-suppressor genes in cancer, were published in the late 1990s [45]. However, these PCR-based approaches are restricted to the study of few candidate genes and are not suitable as screening techniques to identify novel markers. To overcome this, techniques like Amplification of InterMethylated Sites (AIMS) and Restriction Landmark Genomic Scanning (RLGS), which combine the use of methylation-sensitive restriction endonucleases with one-dimensional or two-dimensional electrophoresis were established [46, 47]. These techniques are time consuming, and every new fragment identified as differentially methylated between a control and a test sample has to be cloned and sequenced.
An important step forward was been made in recent years with the introduction of the microarray technology [48], which allowed the simultaneous study of epigenetic changes of thousands of known sequences. Some of these array-based DNA methylation techniques are based on the enrichment of methylated DNA and sequential hybridization onto a dedicated microarray containing thousands of promoters or CpG islands, or even a tiling path array containing the whole genome [49–51]. Enrichment for methylated DNA can be achieved by immunoprecipitation of methylated sequences with an antibody specific for 5-methylcytosine (MeDIP) [52] or by methyl-CpG immunoprecipitation (MCIp), which uses a recombinant protein made of the methyl-CpG-binding domain of the MBD2 protein and the Fc fraction of the human IgG1 to directly isolate methylated DNA [53, 54]. Methylated DNA can also be isolated by digestion with methylation-specific endonucleases [50, 55, 56]. One of the limitations of these methods is that they only provide a blurry picture of the methylome and it is not possible to determine the methylation status of specific CpGs. This problem can be overcome by combining bisulfite treatment of the DNA and microarrays able to differentiate methylated and unmethylated alleles [57–59].
These methods allow a direct detection of DNA methylation patterns. However, there is an additional, but indirect, way for detecting hypermethylated genes. This method applies gene expression profiling before and after treatment with DNA demethylating agents like 5-aza-2′-deoxycytosine (5-AZA) , so that hypermethylated genes become reactivated after treatment [60, 61]. Although this technique has allowed the detection of novel cancer-related hypermethylated genes, 5-AZA is highly toxic to the cells and can alter the expression levels of many genes regardless of their methylation status, leading to a high false-positive and false-negative rates, and a thorough and time-consuming data validation [43].
With regard to histone modifications , global alterations can be detected by isolating histone fractions by high-performance liquid chromatography (HPLC), and then analyzing them by high-performance capillary electrophoresis (HPCE) and liquid chromatography–electrospray mass spectrometry (LC–ES/MS) [62]. Specific modifications at each amino acid residue can also be characterized using antibodies in western blots, immunostaining [63], or tandem mass spectrometry (MS/MS) [62]. If the goal is to detect histone modifications at specific DNA stretches, a different strategy is necessary. DNA can be crosslinked to the associated histones and in a second step precipitated with antibodies specific to certain histone modifications (technique called chromatin immunoprecipitation or ChIP) so that a DNA fraction enriched for that specific histone modification can be isolated. Then, a PCR using primers for the region of interest can be used. Alternatively, a genome-wide picture of histone modifications can be obtained by the ChIP-on-chip technique. By means of this technique, the immunoprecipitated fraction can be labeled and directly hybridized onto a microarray or compared with the input DNA in two-color hybridization [64, 65].
In spite of the potential of microarrays to characterize DNA methylation and histone modifications across the genome, they are limited either by resolution, type and number of sequences analyzed, or by their quantification accuracy. In any case, the complete characterization of the human epigenome of a given sample requires the quantification of the methylation status of each of the ~55 million CpG dinucleotides per diploid cell and the distribution of histone marks of every DNA region, and today this is far from possible using the current microarray platforms. The development of a new generation of sequencers is now revolutionizing both genomics and epigenomics [66–68]. These new sequencing technologies are based on pyrosequencing using millions of picoliter-scale reactions, sequencing by synthesis and sequencing by ligation [66], and are able to sequence up to 2 gigabases of DNA (and the human genome is made of ~3.1 gigabases) in a single experiment. The initial applications of these technologies in the epigenomics field have enabled profiling of the distribution of 20 different histone methylation marks in human CD4+ T-cells [69] or the chromatin state (using six different histone marks) of mouse embryonic stem cells and lineage-committed cells like neural progenitor cells and embryonic fibroblasts [70]. With regard to the detection of DNA methylation changes by high-throug hput sequencers [71], the reduction from 4 to 3 bp of unmethylated sequences (C is transformed to U and then to T in the PCR reaction) after bisulfite treatment poses a methodological problem to identify the origin of the sequenced fragments. Therefore, current technologies do not yet allow direct sequencing of bisulfite-treated whole genomic DNA, although this will be most likely achieved in the near future.
16.4 DNA Hypomethylation in Cancer
One of the initial epigenetic changes discovered in cancer is that, in comparison to normal cells, cancer cells are characterized by a global hypomethylation of the DNA [18, 72, 73]. Such global loss of methylation mainly targets repetitive DNA sequences, coding regions, and introns. So far, it has been reported that DNA hypometh ylation leads to generation of chromosomal instability, reactivation of transposable elements, and loss of imprinting in cancer [74]. Hypomethylation of DNA repeats results in a more open chromatin at those genomic regions and renders the DNA more susceptible to suffer DNA breaks by mitotic recombination of homologous repetitive sequences in different chromosomes (Fig. 16.1) [75–77]. This leads to chromosomal rearrangements, which represent one of the genetic hallmarks of cancer. Also, germline mutations in the DNMT3B gene in humans are associated with the ICF (immunodeficiency, centromeric region instability, facial abnormalities) syndrome, which displays DNA hypomethylation of repeats (like satellite 2), and chromosomal instability [78]. Hypomethylation of DNA in malignant cells can reactivate endoparasitic DNA, such as L1 (long interspersed nuclear elements), and Alu (recombinogenic sequence) repeats [79, 80]. These unmethylated transposons can be transcribed or translocated to other genomic regions so that chromosomal instability is further generated. The loss of methyl groups from DNA can also disrupt genomic imprinting. For instance, the hereditary Beckwith-Wiedemann syndrome shows loss of imprinting of IGF2 (the insulin-like growth factor gene) and individuals affected with this syndrome have an increased risk of cancer [81].
16.5 Gene Silencing in Cancer by DNA Methylation
Tumor-suppressor gene silencing by CpG island hypermethylation is perhaps the best studied epigenetic change in cancer development (Fig. 16.1) [10, 11, 17, 82, 83]. The presence of CpG island promoter hypermethylation affects genes regulating virtually all important cellular functions, such as cell cycle (p16INK4a, p15INK4b, RB1, p14ARF), DNA repair (BRCA1, hMLH1, MGMT, WRN), cell adherence and invasion (CDH1, CDH13, EXT1, SLIT2, EMP3), apoptosis (DAPK, TMS1, SFRP1), carcinogen metabolism (GSTP1), hormonal response (RARB2, ER, PRL, TSH receptors), Ras signaling (RASSF1A, NOREIA), and microRNAs, among others [82]. Table 16.1 shows a summary of the most common hypermethylated genes in human cancer.
Table 16.1
Summary of the best characterized genes silenced by CpG island promoter hypermethylation in human cancer (adapted from [3])
Gene | Function | Location | Cancer type |
|---|---|---|---|
APC | Inhibitor of beta-catenin | 5q21 | Aerodigestive tract |
AR | Androgen receptor | Xq11 | Prostate |
BRCA1 | DNA repair, transcription | 17q21 | Breast, ovary |
CDH1 | E cadherin, cell adhesion | 16q22.1 | Breast, stomach |
CDH13 | H cadherin, cell adhesion | 16q24 | Breast, lung |
COX2 | Cyclooxygenase-2 | 1q25 | Colon, stomach |
CRBP1 | Retinol-binding protein | 3q23 | Colon, stomach, lymphoma |
DAPK | Pro-apoptotic | 9q34.1 | Lymphoma, lung, colon |
DKK1 | Extracellular Wnt inhibitor | 10q11.2 | Colon |
ER | Estrogen receptor | 6q25.1 | Breast |
EXT1 | Heparan sulfate synthesis | 8q24 | Leukemia, skin |
FAT | Cadherin, tumor suppressor | 4q35 | Colon |
GATA4 | Transcription factor | 8p23 | Colon, stomach |
GATA5 | Transcription factor | 20q13 | Colon, stomach |
GSTP1 | Conjugation to glutathione | 11q13 | Prostate, breast, kidney |
HIC1 | Transcription factor | 17p13.3 | Multiple types |
HOXA9 | Homeobox protein | 7p15.2 | Neuroblastoma |
ID4 | Transcription factor | 6p22.3 | Leukemia |
IGFBP3 | Growth-factor-binding protein | 7p13 | Lung, skin |
Lamin A/C | Nuclear intermediate filament | 1q21.2 | Lymphoma, leukemia |
LKB1/STK11 | Serine–threonine kinase | 19p13.3 | Colon, breast, lung |
MGMT | DNA repair of 06–alkyl-guanine | 10q26 | Multiple types |
MLH1 | DNA mismatch repair | 3p21.3 | Colon, endometrium, stomach |
NORE1A | Ras effector homologue | 1q32 | Lung |
p14ARF | MDM2 inhibitor | 9p21 | Colon, stomach, kidney |
p15INK4B | Cyclin-dependent kinase inhibitor | 9p21 | Leukemia |
p16INK4A | Cyclin-dependent kinase inhibitor | 9p21 | Multiple types |
p73 | p53 homologue | 1p36 | Lymphoma |
PR | Progesterone receptor | 11q22 | Breast |
PRLR | Prolactin receptor | 5p13.2 | Breast |
RARB2 | Retinoic acid receptor-beta2 | 3p24 | Colon, lung, head, and neck |
RASSF1A | Ras effector homologue | 3p21.3 | Multiple types |
RB1 | Cell-cycle inhibitor | 13q14 | Retinoblastoma |
RIZ1 | Histone/protein methyltransferase | 1p36 | Breast, liver |
SFRP1 | Secreted frizzled-related protein | 18p11.21 | Colon |
SLC5A8 | Sodium transporter | 12q23 | Glioma, colon |
SOCS1 | Inhibitor of JAK–STAT pathway | 16p13.13 | Liver, myeloma |
SOCS3 | Inhibitor of JAK–STAT pathway | 17q25 | Lung |
SRBC | BRCA1-binding protein | 1p15 | Breast, lung |
SYK | Tyrosine kinase | 9q22 | Breast |
THBS1 | Thrombospondin-1, anti-angiogenic | 15q15 | Glioma |
TMS1 | Pro-apoptotic | 16p11 | Breast |
TPEF/HPP1 | Transmembrane protein | 2q33 | Colon, bladder |
TSHR | Thyroid-stimulating hormone receptor | 14q31 | Thyroid |
VHL | Ubiquitin ligase component | 3p25 | Kidney, hemangioblastoma |
WIF1 | Wnt inhibitory factor | 12q14.3 | Colon, lung |
WRN | DNA repair | 8p12 | Colon, stomach, sarcoma |
Some of the genes hypermethylated in cancer are common to most cancer subtypes, whereas others are considered to be cancer subtype specific [83, 84]. Hierarchical cluster analysis of DNA methylation profiles of tumor suppressor genes in different cancers leads to a classification according to diagnosis and therefore, each cancer type can be assigned a specific DNA hypermethylome. Such patterns of epigenetic inactivation occur not only in sporadic cancers but also in inherited cancer syndromes [85], in which hypermethylation can be the second lesion in the classical Knudson’s two-hit model of cancer development [85, 86].
So far, most of epigenetic studies have targeted known tumor-suppressor genes. However, with the advent of the science of epigenomics, a more precise and less biased delineation of the cancer cell epigenome is becoming accessible. This strategy is already providing a complete new generation of epigenetic markers in cancer. An increasing number of microarray-based studies have focused on the detection of differentially methylated biomarkers associated with specific types of solid tumors like breast cancer [87–90], colorectal cancer [91–94], prostate cancer [95–97], lung cancer [98–101], head and neck cancer [49], oligodendroglioma [102], medulloblastoma [103], and Wilms tumors [104]. In hematological cancers, due to the large number of different cell types of the hematopoietic system, a wide range of different leukemias and lymphomas have been identified by means of morphological, immunohistochemical, and genetic features [105]. Now, several groups are using microarray-based DNA methylation profiling to characterize the epigenome of this heterogeneous group of diseases and to identify diagnostic epigenetic marks. These studies have shown differential methylation profiles between mantle cell lymphoma (MCL) and follicular lymphoma (FL) [106], B-cell chronic lymphocytic leukemia (B-CLL), MCL, and FL [107, 108], cutaneous T-cell lymphoma and normal T-cells [109], acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) [110], AML and normal monocytes [54], and ALL and normal peripheral blood [111]. Taken together, these studies and our recent unpublished experiments indicate that cancer cells, depending on the tumor subtype, might contain up to 2000 hypermethylated gene promoters.
The acquisition of differential methylation in cancer, like hypermethylation of tumor-suppressor genes, is thought to provide the tumor clone with a selective (e.g., proliferative) advantage. However, recent reports have proposed that there is an instructive mechanism behind aberrant DNA methylation in cancer [112, 113]. Keshet and colleagues performed a MeDIP-on-chip study in colon and prostate cancer and as well as identifying differentially methylated genes, they studied whether these genes show distinct biological features [112]. They discovered that genes differentially methylated in cancer are enriched for functional categories (e.g., cell adhesion, cell-cell signaling, signal transduction, and ion transport) and that the expression of some of them is already repressed (or expressed at low levels) in cells from matched normal tissues [112]. Furthermore, they detected a significant enrichment of sequence motifs and a significant clustering of such genes in chromosomal regions [112]. In line with this finding, another study has shown that large stretches of DNA containing several genes can become hypermethylated in cancer [114].
Various independent studies have recently provided further evidence for an instructive mechanism leading to selective methylation of certain groups of genes in most types of cancer. These studies took advantage of ChIP-on-chip data generated using antibodies against members of the polycomb repressor complex 2 in embryonic stem cells [115, 116]. The investigators found that a highly significant proportion of genes that become hypermethylated in cancer were already repressed at the embryonic stem cell stage by PcG marks [117–120]. These findings suggest that epigenetic changes of polycomb target genes occurring in a cell with stem cell features might represent the initial event in tumorigenesis (Fig. 16.2), which supports the cancer stem cell theory [10, 113, 121].
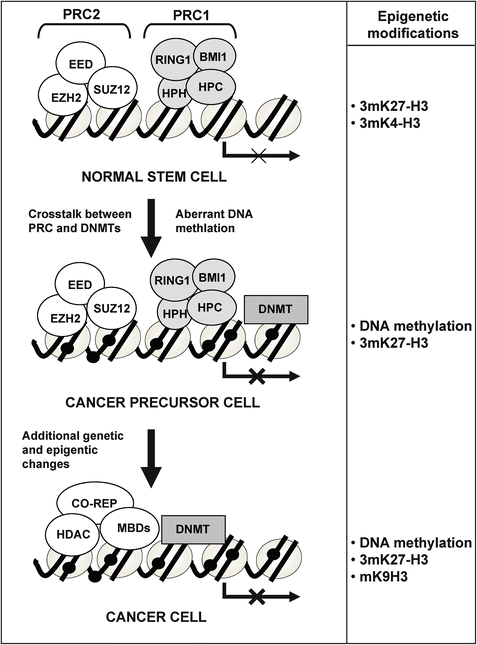
Fig. 16.2
Model of tumorigenesis by which cancer is initiated by a polycomb repressor complex (PRC)-associated DNA methylation followed by additional genetic and epigenetic changes (adapted from [120]).
16.6 DNA Methylation also Targets miRNAs in Cancer
Aberrant repression of gene expression in cancer not only targets coding sequences but also small non-coding RNAs like miRNAs. These are made of short stretches of 22 nucleotides that are able to regulate gene transcription of target genes by sequence-specific base pairing in the 3′ UTR regions and subsequent degradation of the target mRN A or inhibition of translation. Target genes of miRNAs are involved in cellular functions like cell proliferation, differentiation, and apoptosis [122, 123]. Therefore, it is not surprising that miRNA expression has been found to be deregulated in cancer development [124, 125]. The role as tumor suppressors of miRNAs has been investigated in more detail for particular cases. For example, the downregulated let-7 and miR-15/miR-16, and miR-127 are known to target the oncogenic factors RAS and BCL-2, respectively [126, 127]. This may be explained by the failure of these miRNAs during post-transcriptional regulation in cancer cells [128], but additional mechanisms such as CpG island hypermethylation could also be involved. For instance, it has been observed that 5 % of human miRNAs are upregulated by treatment of bladder cancer cells with DNA demethylating agent and HDAC inhibitor [129]. In particular, miR-127 expression was induced by a decrease in DNA methylation levels around the promoter region of the miR-127 gene, and the proto-oncogene BCL6, a potential target of miR-127, was translationally downregulated after treatment [129]. Additionally, using a genetic approach that takes advantage of the genomic disruption of DNMT1 and DNMT3B in cancer cells, we have demonstrated that CpG island hypermethylation is a mechanism that can account for the downregulation of miRNAs in human cancer [130]. Most importantly, the epigenetic silencing of miR-124a, one of the DNA methylation-associated silenced miRNAs isolated using this approach, leads to activation of cyclin D kinase 6 (CDK6), a bona fide oncogenic factor, and phosphorylation of the retinoblastoma (RB1) tumor-suppressor protein [130].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


