Fig. 4.1
Myxopapillary ependymoma, WHO grade I, demonstrates centrally placed hyalinized blood vessels, surrounded by mucinous microcystic degeneration and collars of small epithelioid ependymal cells (courtesy of Dr. A. Perry)
WHO grade II ependymomas consist of four histologic subclasses: cellular, papillary, clear cell, and tanycytic. These tumors are usually solid and well demarcated, with limited infiltration of surrounding structures. Histologic hallmarks include perivascular pseudorosettes, which consist of neoplastic cells encircling a blood vessel with cytoplasmic processes extending between their nuclei and the vessel wall (Fig. 4.2). Less commonly, true ependymal rosettes may form, which consist of neoplastic cells forming a central space reminiscent of ependymal canals. Grade II ependymomas are moderately cellular with low mitotic activity, but may demonstrate nuclear atypia, occasional mitoses, and foci of necrosis and calcification. Interestingly, a review of 134 patients with spinal ependymomas of all grades illustrated a better progression-free survival in patients with WHO grade II ependymomas when compared with WHO grade I ependymomas (Tarapore et al. 2013). Cellular ependymoma is a variant with conspicuous cellularity, but often less prominent pseudorosette or rosette formation. Papillary ependymoma histologically mimics the pattern of choroid plexus papilloma (Louis et al. 2007). Clear cell ependymomas consist of cells with swollen, clear cytoplasm, and well-defined plasma membranes (Louis et al. 2007). This subtype has been shown to demonstrate necrosis and microvascular proliferation with higher clear cell percentage corresponding to higher proliferation (Ishizawa et al. 2012; Fouladi et al. 2003). Tanycytic ependymoma is the rarest ependymoma subtype, occurs mostly within the spinal cord, and is derived from a subpopulation of ependymal cells titled tanycytes. Tanycytic ependymomas tend to contain low-to-moderate cellularity with dense nuclear zones of spindle cells and hypocellular fibrillary zones. These tumors tend to lack the stereotypical rosettes commonly seen in ependymomas, but may contain less organized perivascular rosettes. This subtype appears to show a significant male predominance (Agarwal et al. 2014).
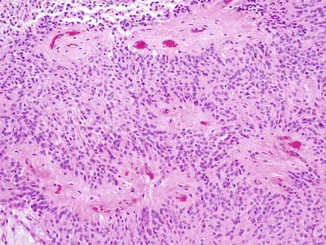
Fig. 4.2
Ependymoma, WHO grade II, demonstrates prominent perivascular pseudorosettes composed of fibrillar nuclear-free zones surrounding centrally placed blood vessels. The mitotic index is low and there is no microvascular proliferation (courtesy of Dr. A. Perry)
WHO grade III anaplastic (malignant) ependymoma has histological evidence of anaplasia, including high cellularity, variable nuclear atypia and hyperchromatism, and marked mitotic activity (Fig. 4.3). Vascular proliferation is often prominent, and necrosis may be widespread (Louis et al. 2007).
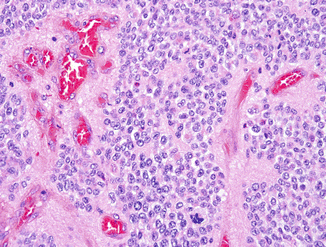
Fig. 4.3
Ependymoma, WHO grade III, demonstrates similar perivascular pseudorosettes to those of lower-grade ependymomas, but also features frequent mitoses and early microvascular proliferation (courtesy of Dr. A. Perry)
Ependymoblastomas are highly malignant rare tumors of embryonic origin that consist of elements resembling primitive embryonic ependymal cells. Despite their name, these tumors are not ependymal tumors, but highly malignant primitive neuroectodermal tumors.
The considerable variation in histopathology among the tumor subtypes of ependymomas can result in discordance among pathologists in grading and diagnosis. The rate of misclassification can be as high as 69 % (Robertson et al. 1998), and the criteria for distinction between grades II and III are not highly reproducible. The WHO guidelines delineate grade II from grade III tumors based on increased mitotic activity, microvascular proliferation, and pseudopalisading necrosis (Louis et al. 2007). Many studies, even with central review, could not confirm the correlation between histology and patients’ outcomes (Ross and Rubinstein 1989; Schiffer et al. 1991; Perilongo et al. 1997; Robertson et al. 1998). Ellison and others completed a histopathological comparison study among five neuropathologists using 229 ependymoma tissue samples. This study found that five out of five concordance on grading based on WHO schema were present only 42 % of the time, and even with consensus, grade was not consistently associated with survival outcomes (Ellison et al. 2011).
Immunohistochemistry (IHC) may be difficult to interpret in ependymomas. IHC varies according to ependymoma subtype and location and may prove to be applicable as a prognostic indicator. GFAP appears to be the most consistent immunohistochemical stain among ependymomas. Multiple studies have shown variable vimentin, synaptophysin, EMA, and S-100 staining among and within ependymoma subtypes and within individual tumors, depending on sampling size (Agarwal et al. 2014; Koperek et al. 2004; Lamzabi et al. 2013; Vege et al. 2000). IHC patterns may also change according to anatomic location of ependymoma. For example, higher expression of neuronal marker, NeuN, appears to occur in supratentorial tumors, whereas GFAP may prove to have higher concentrations in infratentorial tumors (Hagel et al. 2013).
Electron microscopy can be useful in confirming the diagnosis of ependymoma when there is atypical appearance under light microscopy. This is true regarding visualization of rosettes, which are found in more than 90 % of cases, but present in only 30–40 % of cases by light microscopy. Typical ependymoma morphology reveals microvilli and cilia on the apical surface with fragmented microtubules and cellular processes within the intracellular space (Balovannis and Balovannis 2014; Sara et al. 1994). Papillary ependymomas also show a basement membrane, and myxopapillary ependymomas reveal elongated cells within the intracellular space (Balovannis and Balovannis 2014). Higher-grade ependymomas differ from lower grades by lacking cilia and basal bodies (Alfaro-Cervello et al. 2015).
4.2.2 Genetics and Molecular Biology
Ependymomas may arise in any of three anatomic locations including the cerebral hemispheres, the spinal cord, and the posterior fossa. Over 90 % of pediatric ependymomas arise in the brain with approximately 70 % of these cases in the posterior fossa (Mack and Taylor 2009). When ependymomas are examined as a whole, genetic abnormalities have been identified in varying frequencies within the tumors. Chromosomal losses have been described in 1p, 3, 6q, 9p, 10q, 13q, 16p, 17, 21, and 22q. Chromosomal gains have been seen in 1q, 4q, 5, 7, 8, 9, 12q, and 20 (Mack et al. 2009). Abnormalities of chromosome 22 are most often seen including partial loss, monosomy 22, and translocations with a frequency ranging from 26 % to 71 % (Taylor et al. 2005). Loss of NF2, located on chromosome 22, suggests that it is acting as a tumor-suppressor gene in this setting. Indeed, patients with NF2 have a higher incidence of ependymoma (in addition to schwannomas and meningiomas) (Evans et al. 2000). NF2 abnormalities have only correlated with spinal ependymoma and are rare in childhood intracranial ependymoma (Pezzolo et al. 2008). More pertinent to pediatric CNS tumors chromosome 1q gain has been found in up to 22 % of childhood ependymoma (Mack et al. 2009). Gain of 1q is correlated with pediatric age, posterior fossa, high grade, and recurrence (Mendrzyk et al. 2006; Carter et al. 2002; Godfraind et al. 2012). Although these translocations have been informative in describing this disease and patterns of location where it arises, it is important to recognize that a large proportion of pediatric, intracranial disease possesses a balanced comparative genetic hybridization (CGH) profile (Mack et al. 2009).
More detailed studies found specific gene expression patterns and chromosomal translocations in each region (Taylor et al. 2005). Array comparative genetic hybridization (aCGH) recapitulated prior findings such as gains in 1q correlating with pediatric age, intracranial location, and high grade, as well as widespread chromosome 22 loss in intracranial tumors (Taylor et al. 2005; Modena et al. 2006). This high-resolution hybridization study revealed specific losses in chromosome 9p (p16INK4a) and gains in chromosome 7p (TWIST1) and chromosome 20 (Notch pathway signaling) (Taylor et al. 2005; Modena et al. 2006). These analyses reconfirmed results obtained from studies using less sensitive methods suggesting that approximately half of posterior fossa ependymomas have a balanced karyotype (Taylor et al. 2005).
Gene expression analyses demonstrated characteristic networks associated with ependymoma tumors arising from individual sites (Taylor et al. 2005). Ephrin and Jagged signaling pathways and cell cycle mediators (Cdk2, 4 and CyclinB2, D1, G2) are upregulated in supratentorial ependymomas. This is in contrast to spinal cord ependymomas which upregulate Hox transcription factors and IGF signaling and posterior fossa tumors which upregulate ID (bind/inhibit HLH transcription factors) and aquaporin family proteins (Taylor et al. 2005).
Alterations of classic oncogenes associated with other CNS tumors such as astrocytomas and oligodendrogliomas (such as TP53, CDKN2A, and EGFR) are rare in ependymomas (Bijlsma et al. 1995; Ohgaki et al. 1991; Sato et al. 1996). An exception are ErbB2 and ErbB4 which are reported to be overexpressed in the majority of ependymomas (Gilbertson et al. 2002). Clues to a different mechanism of genetic alteration stemmed from work focused on hypermethylation of a p53-dependent transcription repressor, HIC-1 (Waha et al. 2004). Epigenetic methylation acts to transcriptionally repress target genes by placing methyl group on cytosines found within CpG dinucleotides by DNA methyltransferase enzymes (Bird and Wolffe 1999). Decreased expression of HIC-1 is correlated with hypermethylation and astrocytic gliomas, medulloblastomas, and hepatocellular carcinomas in human patient-derived samples (Waha et al. 2004). Of all ependymal tumors analyzed, 83 % showed hypermethylation of HIC–1 which correlated with decreased expression of HIC-1 in 81 % of ependymomas (Waha et al. 2004). Subsequent studies on epigenetic changes employed genome-wide methylation array experiments (Rogers et al. 2011). This approach demonstrated a hypermethylation phenotype in supratentorial and spinal tumors and a hypomethylation pattern in posterior fossa tumors (Rogers et al. 2011). Genes repressed in supratentorial/spinal cord tumors included those involved in cell growth (MAPK10, PPARG) and the immune response (NOD2, IRF7, IRAK3, OSM, PI3) (Rogers et al. 2011).
Microarray expression data from two independent cohorts of posterior fossa ependymomas were able to segregate all tumors into three classifications: supratentorial, posterior fossa and posterior fossa + spinal tumor (Witt et al. 2011). These posterior fossa ependymomas could be further divided into a Group A and Group B (Witt et al. 2011). Whereas Group A tended to occur in younger patients, it was more likely to invade into the cerebellum, had multiple increased cancer-related signaling networks, and had increased recurrence and mortality in 5 years; there was no difference in histology or grade between the groups (Witt et al. 2011). In comparison to Group B, these Group A tumors had a predominantly balanced karyotype with the exception of 1q gain.
Unbiased large-scale methods have led to the discovery of mechanistic differences between ependymoma subgroups. Whole genomic and exomic sequencing showed a strikingly low number of single nucleotide variants (SNV) in both Group A and Group B posterior fossa (PF) ependymomas (Mack et al. 2014). Analysis of the DNA CpG island methylome across all ependymomas yielded three methylome patterns corresponding to supratentorial, posterior fossa, and mixed spinal/posterior fossa (MSPF) tumors (Mack et al. 2014). Posterior fossa Group A (PFA) corresponded to the PF group, and posterior fossa Group B (PFB) tumors corresponded to the MSPF tumors. The methylome of PFA tumors was markedly different from PFB tumors and exhibited significantly higher numbers of CpG hypermethylation, thus conferring a “CpG island methylator” (CIMP) phenotype (Mack et al. 2014). These PFA tumors display a hypermethylation signature, which acts to silence PRC2 target genes. Silencing of these genes prevents differentiation, thus keeping cells in a more “stemlike” program. Most excitingly, treatment of PFA tumors with demethylating agents and repressors of PRC2 was able to restore gene expression signatures. In addition, these agents were active against xenografted PFA tumors and reduce their tumor-initiating capability (Mack et al. 2014).
Overexpression of EZH2 (enhancer of zeste homolog 2) is another recently identified molecular marker that may have prognostic significance. This gene is located along the long arm of chromosome 21 at position 21.2. EZH2 leads to chromatin remodeling by adding a methyl group to histone protein H3K27 and ultimately silencing cyclin-dependent kinases responsible for limiting cell division and pluripotency. Overproduction of EZH2 has previously been implicated in other solid tumors including breast and prostate and other brain tumors such as glioblastoma and atypical rhabdoid teratomas. In the ependymoma population, EZH2 overexpression has been associated with poorer 5-year progression-free and overall survival. This effect appears especially pronounced when combined with AKT (activated protein kinase B) overexpression. Expression of p16 (tumor protein 16) appears to offer a protective role leading to overall improved survival (Li et al. 2015).
4.3 Clinical Features
Data from the Surveillance, Epidemiology and End Results study (SEER) illustrates that nearly half of primary ependymomas occur in the brain and the other half occur within the spinal cord or cauda equina. Spinal cord ependymomas are common in adult patients, although the incidence of ependymomas within the ventricles and cerebellum also increases with age. In children, however, ependymomas occur more frequently in the brain. Histology, too, changes with age; the anaplastic subtype is more common in children and adolescents, while adults are more frequently diagnosed with grade I ependymomas (Villano et al. 2013).
Approximately 35 % of patients have WHO grade III histology at diagnosis. Seven to 15 % of patients with ependymoma have disseminated disease at diagnosis (Perilongo et al. 1997; Robertson et al. 1998). Supratentorial ependymomas grow as intraparenchymal tumors, typically adjacent to the lateral ventricles. Infratentorial ependymomas arise from the fourth ventricle and typically invade adjacent structures or extend into the aqueduct of Sylvius, foramen of Magendie, foramen of Luschka, or to the upper cervical cord. Extraneural ependymomas have been rarely described, usually after progression of intracranial disease. The sites of extraneural spread include peritoneum, lymph nodes, lungs, pleura, bone, and liver (Newton et al. 1992). Extraneural metastases can be seen with both the low- and high-grade ependymoma subtypes (Cimino et al. 2014; Pachella et al. 2015; Perez-Bovet et al. 2013). WHO grade 1 myxopapillary tumors, specifically, have been shown to have a high predilection for metastasis, both within the CNS and in extraneural spaces. Metastatic disease appears to be more common in the pediatric population (Cimino et al. 2014; Fassett et al. 2005). Chang’s staging system for posterior fossa tumors (Table 4.1) can be applied to categorize ependymomas, although this is less commonly used in clinical practice.
Table 4.1
Modified Chang’s staging system for posterior fossa tumors
Definition | ||
|---|---|---|
Tumor | T1 | Tumor confined to the fourth ventricle |
T2 | Tumor of the fourth ventricle with contiguous extension inferiorly through the foramen Magendie and extending to the upper cervical canal | |
T3 | Tumor of the fourth ventricle with lateral extension through the foramen of Luschka into the cerebellomedullary or cerebellopontine cistern | |
T4 | Tumor of the fourth ventricle with invasion of other structures such as the cerebellar peduncle, medulla, pons, midbrain, etc. | |
Metastases | M0 | No evidence of metastases |
M1 | Microscopic tumor found in cerebrospinal fluid | |
M2 | Gross nodule seedings in the cerebellar or cerebral subarachnoid space or in the third or lateral ventricles | |
M3 | Gross nodule seedings in the spinal subarachnoid space | |
M4 | Extraneuroaxial metastases | |
Posterior fossa ependymomas typically present with signs and symptoms of obstructive hydrocephalus including vomiting, headache, and ataxia (Ilgren et al. 1984; Nazar et al. 1990). Infiltration into the brainstem and growth through the foramina of Luschka or central canal may result in cranial nerve palsies, torticollis, or meningismus. Children less than 2 years of age tend to present with nonspecific signs such as irritability, vomiting, lethargy, macrocephaly, or gait disturbance (Nazar et al. 1990). The duration of symptoms is usually less than 6 months at the time of diagnosis (Coulon and Till 1977), with 50 % of children presenting with duration of symptoms of 1 month or less (Horn et al. 1999). Symptoms of spinal cord ependymoma from an adult series included pain in 75 % of patients, sensory changes in 71 %, and weakness in 68 %. The average duration of symptoms was 13 months prior to diagnosis (Waldron et al. 1993). Ependymomas of the cauda equina present with limited spinal motion in 50 % of patients, paravertebral spasm in 32 %, and motor deficits and abolition of reflexes in 34 % of patients (Wager et al. 2000).
4.4 Natural History and Risk Factors
In a small retrospective series of 11 untreated intracranial ependymomas, all patients died within 3 years of symptom onset (Mork and Loken 1977). In an older series, surgery alone was shown to be curative only in a small proportion of patients. For example, in one pediatric series of patients diagnosed between 1935 and 1973, 4/12 patients with intracranial ependymomas and three out of three of patients with spinal cord ependymomas were alive 5 years after surgery (Dohrmann et al. 1976). In another series of patients diagnosed between 1953 and 1974, 2/12 patients with intracranial ependymomas treated with surgery alone were alive 5 years after the surgery, and 10/17 patients with intramedullary ependymomas were alive 10 years after the surgery (Mork and Loken 1977). Another series using better diagnostic imaging indicated that if complete resection was achieved, surgery alone might be curative in a subgroup of children with low-grade intracranial ependymoma. In that study, five out of seven patients treated with gross total resection (GTR) alone remained in remission 24–70 months following surgery (Awaad et al. 1996). Similarly, Hukin et al. reported ten cases of ependymoma treated with complete resection alone. Seven out of ten patients were free of disease and three recurred, with median follow-up of 48 months. Two of the recurrences were salvaged with repeat surgery and radiation therapy (Hukin et al. 1998). Notably, a longer period of observation of patients with ependymoma is necessary, since recurrences continue even after 5 years from diagnosis.
While most studies demonstrate that achievement of GTR of intracranial and intramedullary spinal cord ependymoma correlates with superior outcomes, other risk factors, including location of tumor, histology, and use of adjuvant chemotherapy, have not been unequivocally confirmed to predict outcome (Cervoni et al. 1994; Rousseau et al. 1994; Perilongo et al. 1997; Robertson et al. 1998). Younger children with ependymoma historically have had worse outcomes. It is not clear if age alone, or a combination of risk factors such as unfavorable location, which may preclude GTR, and withholding radiation therapy, may have contributed to poor outcomes in younger age groups. Current therapeutic studies use age, histology, and location of ependymoma for stratification of treatment, despite conflicting literature reports on their validity as prognostic factors.
4.5 Diagnosis
Evaluation of a patient with ependymoma should include a comprehensive history and physical examination, pre- and post-operative magnetic resonance imaging (MRI) of the brain, MRI of the spine, and cerebrospinal fluid evaluation. The spine MRI should ideally be performed prior to surgery, because post-operatively, blood in the spinal subarachnoid space may be confused with drop metastases.
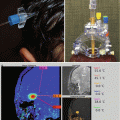
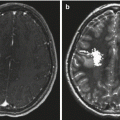
Radiographically, supratentorial ependymomas appear as large, heterogeneous, periventricular, or, less commonly, intraventricular masses. Calcifications are present in approximately 50 % of tumors examined by computerized tomography. Most supratentorial ependymomas have cystic components and enhance after the administration of intravenous contrast (Furie and Provenzale 1995). Infratentorial ependymomas appear as heterogeneous lesions that grow into the fourth ventricle and cause dilation of its upper part. Usually, the tumor is separated from the vermis by a cleavage plane. In most cases, the solid part of the tumor is intense with gray matter on T1- and T2-weighted MR images, and this enhances with contrast (Tortori-Donati et al. 1995) (Figs. 4.4, 4.5, and 4.6).
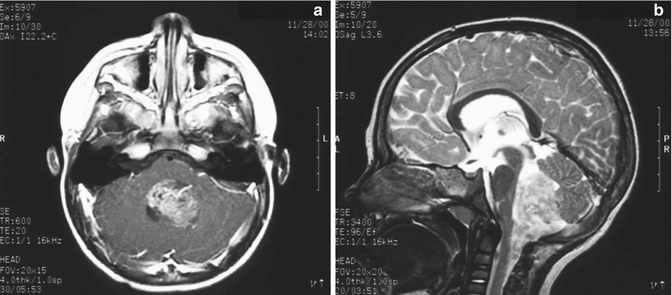
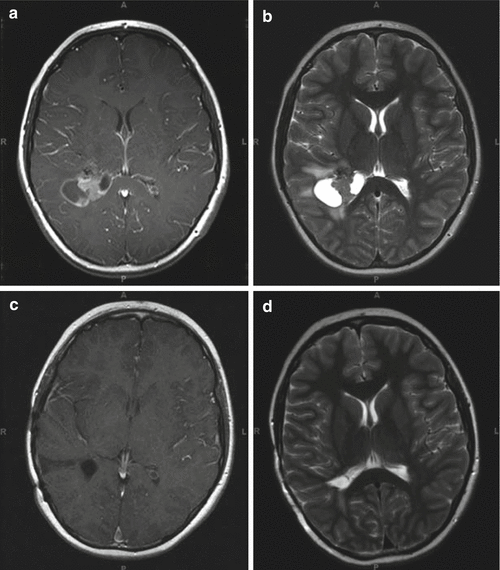
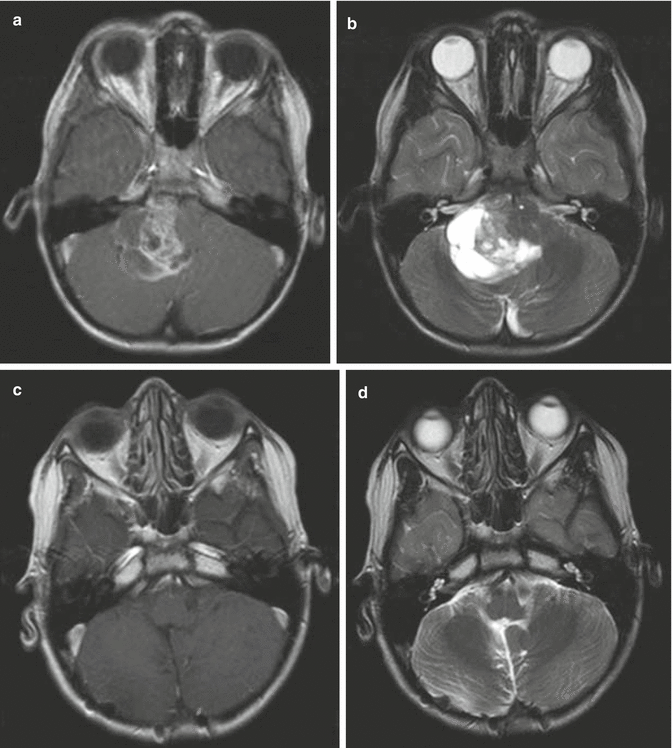
Fig. 4.4
Typical imaging features of a posterior fossa grade II ependymoma showing heterogeneous enhancement following gadolinium administration (a). The T2-weighted image (b) shows the typical extension of the tumor through the foramen magnum into the upper cervical spinal canal
Fig. 4.5
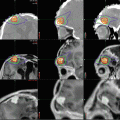
Preoperative T1- (a) and T2-weighted (b) MR images of a supratentorial ependymoma in a 9-year-old boy arising adjacent to the right trigone and periventricular area. The grade III ependymoma was grossly resected and after external beam radiation therapy, he remains progression-free for 3 years. The postoperative T1-weighted image with contrast (c) and T2-weighted image (d) show no evidence of tumor recurrence
Fig. 4.6
A preoperative T1-weighted MR image with contrast (a) and T2-weighted image (b) from a 7-year-old boy with a right cerebellopontine angle grade 2 ependymoma. After resection and external beam radiation therapy, he remains progression-free for 5 years. The postoperative T1-weighted image with contrast (c) and T2-weighted image (d) show no evidence of tumor recurrence
The typical picture of an intramedullary ependymoma on MRI consists of segmental or diffuse cord expansion with intramedullary intensity abnormalities and prominent nodular gadolinium enhancement (see Fig. 10.2). Intramedullary cysts and hydrosyringomyelia are common, particularly in childhood cases. Gadolinium enhancement may distinguish solid tumor from cord edema and from cyst or syrinx (Slasky et al. 1987). Cauda equina ependymomas usually demonstrate homogeneous hypointense signal on T1-weighted MRI sequences, hyperintense signal on T2-weighted sequences, and homogeneous enhancement after gadolinium injection (Wager et al. 2000).
4.6 Treatment
4.6.1 Surgery
The goals of surgery are to make a definitive tissue diagnosis, achieve a GTR of the tumor, and re-establish cerebrospinal fluid flow. The location of the ependymoma may be a limiting factor to surgical resection, particularly when within the upper spinal cord or posterior fossa and abutting midbrain structures (Safaee et al. 2014).
Based on retrospective studies, the extent of surgery is considered as the most important prognostic factor. In a retrospective analysis of 96 pediatric posterior fossa ependymomas from the Children’s Oncology Group, extent of resection and older age were significantly correlated with better overall survival. They went on to review 1,444 patients from 32 manuscripts from 1990 to 2005 and determined that the extent of resection was a significant factor in 21 studies, age in 12, and histological grading in 9 of the studies (Tihan et al. 2008). In a study of 55 patients with anaplastic ependymoma, patients with a complete surgical resection followed by other treatments had an 83 % disease-free survival after 3 years of follow-up, compared with 38 % for those without complete resection (Timmermann et al. 2000). In a similar study of 32 children with ependymoma between 2 and 18 years of age, 66 % of patients with complete resection had 5-year progression-free survival, compared with 11 % of those without complete resection (Robertson et al. 1998).
In patients where complete resection may be unobtainable, some groups have investigated the use of a “second-look surgery.” An Italian group investigated this approach in a population of 38 patients who underwent repeat surgery for a variety of indications: complications impeding resection on first attempt, missed tumor tissue, or anatomic areas where initial resection was not possible. Out of the 38 investigated patients, 21 were able to achieve total resection after one or more repeat surgeries. Many of these patients also received adjuvant chemotherapy or radiation prior to repeat resection attempts. Overall, patients who were ultimately able to achieve complete resection even after multiple surgical attempts still went on to have higher rates of survival when compared to patients that continued to have evidence of disease (Massimino et al. 2011). This study highlights the significance of surgical resection in ependymoma treatment.
A recent review looked at 26 patients with intracranial ependymomas that underwent surgery alone and achieved GTR. This study also evaluated status of outcome in groups of patients with varying Ki-67 indices and status of EZH2 positivity. From this population of 12 posterior fossa and 14 supratentorial tumors, the 5-year overall survival was 70 % in both populations. Patients with higher Ki-67 indices and positive EZH2 status were associated with poorer rates of progression-free survival, despite GTR. These studies outline the potential for risk stratification of ependymoma subtypes from a WHO grade, histological, and ultimately from a molecular standpoint.
Some investigators have used surgery alone to treat ependymoma. In one study of ten selected patients with intracranial ependymoma (eight supratentorial and two posterior fossa tumors), seven remained free of disease without any other interventions at a median follow-up of 48 months (Hukin et al. 1998). Investigators at St. Jude Children’s Research Hospital reported 10/16 patients with residual disease after the first surgery achieved GTR after a “second-look” surgery (Osterdock et al. 2000).
Posterior fossa ependymomas frequently arise from the floor of the fourth ventricle or the region of the foramina of Luschka, extending out to the cerebellopontine angle (Fig. 4.6). Cranial nerves and vascular structures are frequently encased or displaced by the tumor, making surgical excision difficult. Sequelae of an aggressive surgical approach often include cranial neuropathy, bulbar dysfunction, and the risk of posterior circulation infarction. Surgical results have improved through technical advances, such as intraoperative electromyographic monitoring of cranial nerves, computer-assisted navigation, and the operating microscope. Due to the strong prognostic impact of the degree of surgical resection, all children with suspected ependymoma should be referred to a center that can provide the expertise required for optimal management of these tumors.
4.6.2 Radiation Therapy
Radiation therapy (RT) is considered the standard adjuvant treatment of intracranial ependymomas in older children, with different definitions of lower age limit in different studies. The approach to radiation therapy of ependymoma, in particular the radiation field, has changed over a period of decades. In the 1960s and 1970s, local field radiation was used, as improved outcomes were noted in irradiated patients, when compared to historical controls. In 1975, Salazar recommended extending the radiation field to include the whole brain in patients with low-grade ependymomas and the entire craniospinal axis in patients with high-grade ependymomas (Salazar et al. 1975). These recommendations were based on an overestimated risk of dissemination from autopsy findings and on poor distinction between ependymomas and ependymoblastomas. In the late 1980s and early 1990s, multiple studies questioned the role of craniospinal radiation (Shaw et al. 1987; Goldwein et al. 1991; Vanuytsel and Brada 1991), as there was no difference in failure rates between patients undergoing localized versus craniospinal radiation. Local relapse was confirmed to be the most significant component of failure, and a local radiation dose of more than 4,500 cGy was recommended (Goldwein et al. 1991).
In the late 1980s, cooperative pediatric cancer groups initiated studies of postoperative chemotherapy in infant brain tumors with the goal to delay and possibly avoid radiation in young patients for whom large radiation fields would cause significant morbidity (Duffner et al. 1993; Geyer et al. 1994). Young children with ependymomas were treated in these studies together with patients with primitive neuroectodermal tumors. In the 1990s, attempts were made to replace radiation therapy with high-dose chemotherapy consolidation in children less than 6 years of age with malignant brain tumors (Mason et al. 1998) or to replace radiation with postoperative chemotherapy in children under 5 years of age (Grill et al. 2001). However, radiation was avoided in only 23 % of patients in the later study, and after progression of tumor, only those children who underwent a second complete resection remained in remission following radiation therapy.
Hyperfractionated radiation therapy was investigated by the Society for Pediatric Oncology. They reported on 24 children over the age of 5 years, treated with either 60 Gy (for patients with complete resection) or 66 Gy (for patients with incomplete resection), given in two daily fractions. Five-year overall survival was 74 % and progression-free survival was 54 %. The study concluded that although the treatment was well tolerated, the survival figures were comparable to those for more conventional treatment regimens, and hence hyperfractionated therapy did not warrant further investigation (Conter et al. 2009). Other investigators have focused on reducing the radiation field by using conformal radiotherapy. In one such study, 36 children with localized ependymoma underwent conformal radiotherapy with an anatomically defined clinical target volume margin of 10 mm surrounding the postoperative residual tumor and tumor bed. Two failures occurred after a median follow-up period of 15 months. It is of significance that 30/36 children in this preliminary report had complete surgical resection (Merchant et al. 2000). More recently, the authors have published a follow-up study of 153 pediatric patients with ependymoma (median age 2.9 years). Patients received conformal, focal radiation to a dose of 54–55.9 Gy following definitive surgery. Thirty-five subjects had prior chemotherapy. Seven-year local control, event-free survival, and overall survival were 87, 69, and 81 %. Survival was affected by tumor grade and extent of resection (Merchant et al. 2009). Radiosurgery has also been used in patients with ependymoma, usually at the time of recurrence, and it can be used safely without significant risk of radionecrosis (Hodgson et al. 2001). In another study, tumor control was achieved in three out of five patients undergoing radiosurgery for residual localized ependymoma (Aggarwal et al. 1997). It is possible that radiosurgery will have a significant role in local control of ependymoma in the future; however, more studies comparing radiosurgery to standard radiation are necessary.
Given the limited effectiveness of chemotherapy in avoiding radiation (see Sect. 4.6.3) and concerns about radiation-induced neurocognitive deficits, 88 patients at St. Jude’s between the ages of 2.85 and 4.5 years were treated with conformal radiation therapy (54 or 59 Gy) to gross tumor volume plus a margin of 1 mm (Merchant et al. 2004). Median follow-up was more than 3 years. The 3-year progression-free survival rate in this study was nearly 75 %, with a cumulative incidence of local failure as a component of failure (distant + local) at 3 years being 14.8 %. Serial neurocognitive evaluations performed until patients were 24 months postcompletion of radiation therapy revealed stable IQs. In a follow-up study, the group at St. Jude’s reported on outcome of 153 patients with age range of 0.9–22.9 years, median 2.9 years, with localized ependymoma treated with conformal radiation after definitive surgery. Seven-year local control, EFS, and OS were 83.7 %, 69.1 %, and 81.0 %, again supporting the use of conformal techniques in this setting, even for very young patients (Merchant et al 2009). This significant study has prompted continued evaluation of conformal radiotherapy in patients older than 12 months of age, an ongoing Children’s Oncology Group clinical trial to use conformal radiation therapy in a subgroup of patients older than 1 year of age with higher-risk localized ependymomas.
There remain patients, however, that may not benefit from RT. In one retrospective analysis of 92 patients with supratentorial WHO grade I or II ependymomas who underwent GTR with or without associated RT, those that underwent resection alone showed no worse overall survival (83.2 % vs. 84.1 %) when compared to the GTR + RT group (Ghia et al. 2013). In these instances, conservative approaches without radiation therapy may be indicated as long as GTR is achievable, but improved delineation of prognostic features is necessary.
Craniospinal radiation is still to be used alone or in combination with chemotherapy for older patients with disseminated ependymoma.
In summary, although the role of radiation therapy has not been confirmed in randomized studies, the high risk of relapse in younger children treated with chemotherapy only, even after GTR, warrants using this modality. Studies are underway to confirm the role of conformal field radiation in local control of ependymoma.
4.6.3 Chemotherapy
Ependymomas have variable sensitivity to chemotherapy. In multiple studies involving adults with relapsed ependymoma, cisplatin-containing regimens have yielded superior response rates to alternate approaches. Complete and partial responses have been observed at rates that range between 30 (Walker and Allen 1988; Brandes et al. 2005) and 60 % (Gornet et al. 1999). Despite these high rates of response, no chemotherapy regimens have yet been demonstrated to improve overall survival of adults with recurrent ependymoma.
In newly diagnosed patients with ependymoma, chemotherapy has been evaluated for the treatment of children with residual disease or to avoid radiotherapy in young children (<3 years old). In eight children under the age of four with residual ependymoma, an 86 % response rate to VETOPEC therapy (vincristine, etoposide, cyclophosphamide, cisplatin, carboplatin) was reported (White et al. 1998). The Children’s Cancer Group protocol (CCG-9942) investigated the role of preirradiation chemotherapy with vincristine, etoposide, cisplatin, and cyclophosphamide in children older than 3 years of age with residual disease. In a preliminary report from this study, the event-free survival did not differ between patients without residual disease whose postoperative treatment consisted of radiation therapy alone (62 ± 8 %) and patients with residual disease who received chemotherapy and irradiation (55 ± 9 %). The chemotherapy objective response rate was 58 %, but 14 % of patients experienced tumor progression while receiving chemotherapy prior to irradiation (Garvin et al. 2004).
The role of chemotherapy in delaying or avoiding radiation in young children with ependymoma has been well studied (Table 4.2). The outcomes are difficult to compare due to differences in the use of radiation. In the earlier studies, although radiation was planned, it was not always given (Duffner et al. 1993; Geyer et al. 1994). In later studies, radiation was used only after tumor progression, and its use indicated chemotherapy failure (Mason et al. 1998; Grill et al. 2001; Grundy et al. 2007). These issues notwithstanding, several valuable observations regarding the effectiveness of chemotherapy can be made. Grundy et al. report the most favorable outcome data with chemotherapy (vincristine, carboplatin, cisplatin, cyclophosphamide, and methotrexate) in the treatment of 80 children less than 3 years old with non-metastatic intracranial ependymoma (Grundy et al. 2007). The 5-year cumulative incidence for freedom from radiotherapy was 42 % in this group of patients. Further, with a median follow-up of 6 years, this group achieved an overall survival rate of approximately 80 % at 3 years and 60 % at 5 years. While the study is limited by lack of radiographic data and neurocognitive follow-up (Bouffet et al. 2007), it suggests that intensive chemotherapy may have benefit in the treatment of young children with ependymoma. While response in this recent study is promising, there are no data to suggest that the neurocognitive outcome for these patients will be superior to that observed with conformal radiotherapy (Merchant et al. 2004). Thus, concerns about response and outcome have resulted in a reexamination of local radiation in all but the youngest patients in several ongoing clinical trials for ependymoma.
Table 4.2




Progression-free survival of young children with ependymoma treated with chemotherapy
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
