FIGURE 130-1. A, 10% hematoxylin and eosin stain of a peritoneal lesion of endometriosis. Note the presence of glands and stroma surrounded by fibrosis. B, 10% hematoxylin and eosin stain of late proliferative endometrium taken from the same patient at the time of surgery.
Epidemiology
Endometriosis occurs primarily during the reproductive years and is more prevalent in women with few pregnancies (relatively more menstrual cycles) and higher endogenous estrogen levels. Its prevalence is lower in women with decreased estrogen levels and fewer menstrual cycles, such as women with hypothalamic amenorrhea. Supporting data from the Nurses’ Health Study II13–15 demonstrate that increased risks to developing endometriosis include menarche at younger than 10 years of age, low birth weight (<5.5 lbs), body mass index (BMI) over 25, nulliparity, in utero exposure to diethylstilbestrol (DES), a positive family history of endometriosis, and consumption of more than 100 g/day of ethanol. The disorder is greater among Caucasian and Asian > African American > Hispanic women. Lower risk of developing endometriosis is associated with lactation for more than 23 months, parity of more than 3 children, and smoking. No association was found with regard to height, waist/hip ratio, caffeine, or hair color.13–15
Pathogenesis
The first recorded description of pathology consistent with endometriosis was by Shroen in 1690.16,17 It was further described by von Rokitansky18 over 100 years ago, and despite these early descriptions, the precise pathogenesis of this enigmatic disorder remains unknown. Theories about the development of endometriosis fall into two classifications19: those proposing that endometriosis implants arise from uterine endometrium and those proposing that implants arise from tissues other than the endometrium.
ENDOMETRIAL ORIGIN
Benign Metastasis
The theory of benign metastasis holds that ectopic endometrial lesions result from lymphatic or hematogenous dissemination of endometrial cells.20,21,23 Microvascular studies demonstrate lymph flow from the uterus into the ovary, suggesting a possible role for the lymphatic system in the etiology of ovarian endometriosis.24 Endometriosis within lymph nodes has been documented in 6.5% of women at lymphadenectomy and 6.7% of women at autopsy.25 The strongest evidence for the theory derives from reports of histologically proven endometriotic lesions occurring in sites distant from the uterus or pelvis, including bone, brain, and lung.26
Retrograde Menstruation
In menstruating women, approximately 90% of menstrual blood emerges through the cervix, and about 10% is refluxed through the fallopian tubes into the peritoneal cavity. Sampson’s theory of retrograde menstruation and transplantation postulates that sloughed endometrial tissue, fluxed through the fallopian tubes, contains viable, steroid-sensitive cells that are capable of implanting outside the uterus.22 Although this hypothesis is attractive, it does not explain why retrograde menstruation, a physiologic event,27 does not result in endometriosis in all women. The volume of regurgitated menstrual debris has been considered to be relevant to establishment of the disorder, since women with endometriosis have a higher volume of refluxed menstrual blood than those without the disorder.27 The findings of hypotonia of the uterotubal junction in women with endometriosis, but not in controls,28 supports this theory and may in part explain why only some women are affected. In addition, the occurrence during the menstrual phase of subendometrial myometrial contractile waves exhibiting a retrograde pattern in women with endometriosis—which does not occur in controls with a normal antegrade pattern29—further supports Sampson’s theory and suggests that endometriosis may have some of its origin in myometrial dysfunction. The greatest support for Sampson’s theory derives from the nonhuman primate, where spontaneous endometriosis was found in approximately 25% of baboons, and with increasing prevalence with increasing duration of captivity, lack of conception, and incidence of retrograde menstruation.30 In addition, endometriosis can be induced in baboons by cervical occlusion,31 and endometriosis is uniformly observed in adolescent girls with primary amenorrhea caused by cervical hypoplasia or aplasia, vaginal agenesis, or other müllerian anomalies associated with obstruction to the outflow of menstrual blood.32 Even subtle compromise of antegrade menstruation may predispose to endometriosis, as evidenced by a recent report of a higher prevalence of endometriosis in women with a uterine septum.33
A murine model of endometriosis has provided new insight into the pathogenesis of peritoneal endometriosis.34 Conditional activation of the K-ras oncogene in endometrial cells deposited into the peritoneum resulted in histologically confirmed peritoneal endometriotic implants in nearly 50% of mice within 8 months. However, similar activation of the K-ras oncogene in peritoneal cells showed no progression to endometriosis. These results in a mouse model would seem to support the theory of retrograde menstruation in the development of peritoneal endometriosis.
NONENDOMETRIAL ORIGIN
The theory of metaplasia of the coelomic epithelium proposes that normal peritoneal tissue (mesothelium) transforms via metaplastic transition to ectopic endometrial tissue.19,35 The closely related induction theory holds that an endogenous inductive stimulus (hormonal, immunologic) promotes differentiation of undifferentiated cells in the mesothelium into endometrial cells.19 Both theories are based on the common embryologic origin of endometrial and mesothelial cells and imply the presence of incompletely differentiated cells in the mesothelium that are capable of such transformation. Evidence supporting this assumption comes from demonstration of totipotent mesothelial serosal cells in the coelomic lining36 and from co-culture experiments of ovarian surface epithelium with endometrial stromal cells exposed to supraphysiologic levels of 17β-estradiol that resulted in endometriosis arising via a metaplastic transformation of the mesothelium.37 Furthermore, the theory of embryonic müllerian rests postulates that cells residual from embryologic müllerian-duct migration maintain the capacity to develop into endometriotic lesions under the influence of estrogens.38 Clinical reports in support of these theories derive from histologically confirmed endometriotic tissue in patients without menstrual endometrium. For example, endometriosis has been documented in a patient with Rokitansky-Küster-Hauser syndrome (notable in that the patient did not have functioning endometrium).39 Perhaps the most compelling evidence for a nonendometrial etiology comes from cases of men with prostate cancer undergoing high-dose estrogen treatment who were subsequently diagnosed with endometriosis.40,41
Of all these possibilities, the one that has the greatest support is the theory of retrograde menstruation resulting in endometrial tissue that may implant on peritoneal or abdominal/pelvic visceral surfaces. In addition, the tissue may be transported to distant sites by vascular or lymphatic channels.42 Implicit in this theory is that desquamated endometrial tissue contains viable cells capable of implanting in heterologous tissue.
IMMUNE SYSTEM
There is abundant evidence demonstrating that endometriosis is accompanied by an inflammatory reaction in the peritoneum, resulting in abnormal levels of cytokines and chemokines in the peritoneal fluid. There have been observations of higher levels of activated macrophages and cytokines in peritoneal fluid from women with endometriosis than in those without the disorder.43,44 In addition, there is some evidence to suggest a role for abnormal macrophage activity in endometriosis, which is supported by numerous studies showing altered macrophage-dependent cytokine levels and activity that can affect the survival and growth of ectopic endometrial cells.45,46 In addition to inflammation, a growing body of evidence supports relative immune dysfunction in women with endometriosis.46,47 It has been suggested that endometriosis is part of an autoimmune disorder,48 on the basis of observations that women with endometriosis have increased generalized B-cell activity49 and an association with abnormal autoantibodies, such as autoantibodies to phospholipids50 and to endometrium.51 In addition, an increased association with other autoimmune phenomena such as premature ovarian failure and atopic disease supports the hypothesis.52 The actual significance of the increased multiple antibodies remains unknown, however, and the mechanisms by which such an increase in antibodies leads to the development of endometriosis remains uncertain. The possibility exists that the increased antibody production is the consequence of a general inflammatory response initiated by activated macrophages and other antigen-presenting cells in response to the ectopic endometrial tissue, rather than the cause of the development of endometriosis. Before an immune basis for endometriosis can be determined with certainty, well-controlled prospective studies need to be conducted. The role of the humoral and cellular immune systems in the pathophysiology of endometriosis is presented later in the section on pathophysiology.
GENETICS
A genetic predisposition to the development of endometriosis has been recognized.53–56 An increased risk (5% to 8%) of endometriosis is seen among first-degree relatives of affected women in comparison to the general population.57,58 Recurrence risks of this magnitude are most consistent with a polygenic/multifactorial etiology. Additional support for a polygenic inheritance is the earlier age of onset for familial versus nonfamilial cases and similar age of onset among affected relatives.59 In addition, a strong familial tendency is found in nonhuman primates.60 A genetic contribution was recently questioned by Di and Guo61 because of issues with study designs, small sample sizes, ascertainment bias, increased opportunity to diagnosis family members of cases versus controls, and familial aggregation of confounding risks such as early age at menarche. However, large studies in Australian twins,62 the Icelandic population,63 rhesus macaques,64 and the significant linkage to chromosome 10 found in a large collaborative study of 1176 families65 address many of these concerns and together provide strong evidence for a genetic contribution to the disease.
ENVIRONMENT
The perinatal, postnatal, peripubertal, and adult periods are all susceptible windows during which inappropriate exposure to xenoestrogens and other endocrine-disrupting chemicals (EDCs) and other factors can induce developmental programming and increase risk for female reproductive-tract disorders, including endometriosis.66–69 Exposures to EDCs during adulthood, with or without prior exposure, may also predispose to developing or promoting endometriosis.68,69 Monkeys exposed to total body proton irradiation have an increased prevalence of endometriosis when compared with controls (53% versus 26%),70 and irradiation may contribute to the establishment and/or growth of endometriosis.71,72 Also, the evidence is overwhelming in adult nonhuman primates and rodents that endometriosis can be promoted by many organochlorines, including tetrahydrochlorodibenzo-p-dioxin (TCDD), pesticides such as methoxychlor and DDT, and polychlorinated biphenyls.73–75 Severe endometriosis developed in adult rhesus monkeys exposed TCDD at 5 to 25 parts per trillion (ppt) daily for 4 years, and the severity of the disease was dose dependent.73 Also, when adult mice are exposed to TCDD and dioxin-like compounds with subsequent implantation of human endometrial tissue into the peritoneum, the estrogen level and exposure timing demonstrate significant postimplantation endometrial growth in mice and rats.76,77 Furthermore, Nayyar et al.91 reported recently that when adult mice are exposed to TCDD followed by peritoneal seeding of human endometrial tissue, there is lack of progesterone regulation of matrix metalloproteinases (MMPs) in this peritoneal endometriosis model.78
While the animal data are compelling, data linking organochlorine exposure and endometriosis in adult women are equivocal. There are 14 human studies of endocrine disruptors and endometriosis (12 case-control and 2 retrospective cohort studies) evaluating dozens of congeners and with largely inconsistent findings.79,80 Epidemiologic studies demonstrate that Belgium, a country with one of the highest levels of dioxin pollution in the world, has the highest incidence of endometriosis and prevalence of severe disease.79 In 1976 in Seveso, Italy, a cohort of women was acutely exposed to dioxin and evaluated for endometriosis 20 years later. The study demonstrated a doubled, nonsignificant risk for endometriosis among women with serum TCDD levels of 100 ppt or higher; however, no clear dose response was found.80 A more recent case-control study carried out on Italian and Belgian women of reproductive age, with and without endometriosis, showed no significant differences in dioxin-like compound body burdens between women with and without endometriosis.81 The weaknesses of these observational epidemiologic studies include limited sample sizes and confounding variables.
However, data from the Nurses’ Health Study II linking in utero exposure to DES and development of endometriosis in adult women are compelling.14 This study was a prospective study of 116,678 female nurses aged 25 to 42 years old, who filled out a baseline questionnaire in 1989 and were followed at 2-year intervals. The prevalence of endometriosis at baseline was 5%. The incidence was 2941 laparoscopically confirmed cases, with pain symptoms prompting diagnosis in 77% of subjects with endometriosis and an infertility work-up prompting diagnosis in the remaining 33%. A history of in utero exposure to DES increased by 80% a woman’s chance of having laparoscopically proven endometriosis in this cohort. Other risk factors were low birth weight and early menarche, consistent with prolonged exposure and perhaps multiple exposures (in utero and subsequently peripubertal) that increased risk of developing this disorder.14
GENOMIC ALTERATIONS
Alterations in DNA copy numbers in human endometriosis lesions have been detected by comparative genomic hybridization.82 In addition, a recent study using a similar approach revealed several regions of genomic alteration in eutopic endometrium from women with endometriosis compared to normal controls. Some of the regions were the same as previously found in the endometriotic lesions, suggesting that genomic alterations may be a proximate cause for endometriosis.83 Two-color fluorescence in situ hybridization was used for analysis of endometriotic and normal archival tissue, and increased heterogeneity of chromosome 17 aneuploidy in endometriosis was found. These findings support a multistep pathway involving somatic genetic alterations in the development and progression of endometriosis.84
Pathophysiology
For tissue to implant and thrive in an ectopic location, it must survive after detachment from its original location, attach itself and invade into a new environment, proliferate, and establish a new blood supply. These processes involve inhibition of apoptosis; cell-cell and cell-substratum interactions; matrix degradation, inhibition, and repair; controlled cellular proliferation; and angiogenesis. As with normal endometrium, endometriosis tissue responds to cyclic changes in steroid hormones by proliferation and glandular secretion, as well as by production of autocrine and paracrine factors that affect these processes.85
CELL SURVIVAL/CELL ADHESION
Endometrial cells undergo apoptosis as part of their natural cell death at the time of menstruation.86 In women with endometriosis, however, the percentage of sloughed endometrial cells undergoing apoptosis is greatly reduced, thereby resulting in increased numbers of cells that may survive in ectopic sites.87 After survival, endometrial cells must adhere to the mesothelium or other surfaces and then begin the process of invasion. An in vitro model of endometriosis using confocal microscopy has demonstrated the adherence and invasion of endometrial cells through peritoneal mesothelium.88 The expression of cellular adhesion molecules by endometriotic lesions has been investigated to help understand mechanisms involved in the maintenance of endometrial tissue in ectopic locations.89 Of interest is the finding that refluxed endometrial cells, but not tissue fragments, down-regulate expression of the E-cadherin cell adhesion molecule.90 It is possible that proteases, cytokines, or growth factors in the peritoneal fluid of women with endometriosis alter some endometrial structures and may result in their selective adhesion to the peritoneum.
MATRIX DEGRADATION/INVASION
Endometriosis, a benign disorder, can invade tissues and surfaces with as much aggression as a malignancy. Matrix metalloproteinases (MMPs) and their inhibitors (tissue inhibitors of metalloproteinases [TIMPs]) are involved in extracellular matrix remodeling and have been implicated in the endometrial remodeling that occurs during the proliferative phase of the menstrual cycle and as participants in tissue desquamation at the time of menses.91,92 The balance between MMPs and TIMPs is critical in maintaining the appropriate level of MMP activity, and failure to maintain this balance may contribute to matrix breakdown and cellular invasion, as observed in endometriosis.
In eutopic endometrium, mRNA for all the MMPs has been detected during the menstrual phase.91,93 Matrilysin (MMP-7) mRNA is localized to the epithelium, whereas all others are expressed in the stroma. Only MMP-2 and TIMP-1 mRNA is constitutively expressed throughout the cycle. MMP-10 and MMP-9 are expressed only in late secretory and menstrual endometrium, whereas MMP-7, MMP-2, and MMP-11 mRNA is consistently detected in proliferative endometrium, which suggests steroid regulation of MMPs in this tissue. This theory is supported by experimental studies in vitro.92,94,95
Epithelial-specific MMP-7 is expressed in endometriotic lesions during the secretory phase, whereas it is absent in eutopic endometrium,96 which suggests that progesterone suppression of MMP-7 is dysregulated in endometriosis and that this MMP may play a role in growth or progression of the disorder. MMP-3 has also been identified in lesions of endometriosis, and such expression is diminished after danazol treatment.97 MMP-1 is expressed focally in red peritoneal and ovarian endometriosis lesions, regardless of the menstrual phase, but not in black peritoneal and rectovaginal lesions.98 Foci of MMP-1 expression closely correlate with matrix breakdown and with the absence of progesterone receptors in adjacent epithelial cells, thus suggesting that MMP-1 expression may be involved in tissue remodeling and bleeding and possibly reimplantation of endometriotic lesions. An endometriosis-specific protein, ENDO-II, has homology with TIMP-1.99 Both eutopic and ectopic tissue from women with endometriosis exhibit patterns of altered MMP regulation in vivo. A lack of responsiveness to progesterone was demonstrated in vitro, associated with a failure to suppress MMP expression and an enhanced ability of the tissue to establish experimental endometriosis. In vitro treatments with retinoic acid and transforming growth factor β (TGF-β), however, restored the ability of progesterone to suppress MMPs in vitro and prevented the establishment of experimental disease.100 In vitro studies using endometrial cell cultures suggest that interleukin-8 (IL-8) increases MMP activity and the invasive capability of endometrial stromal cells in culture.101 Studies have also shown that MMP expression in human peritoneal endometriotic lesions are much greater than that in eutopic endometrium.102 Specifically, endometriotic tissue possesses higher levels of gelatinase activity than eutopic endometrium from patients with endometriosis, and MMP-9 may be of importance for the implantation and invasive growth of endometriotic tissue that may lead to endometriosis.103 Uterine endometrium from women with endometriosis expressed higher levels of MMP-2 and membrane type (MT)1-MMP and lower levels of TIMP-2 than endometrium from women without disease.104
The hypogonadal, athymic nude mouse is a useful model to investigate steroid dependence of MMPs in the pathogenesis of endometriosis.105 Endometrial explant cultures treated with estradiol secrete MMPs and establish ectopic peritoneal lesions when injected into recipient animals. Suppressing MMP secretion with progesterone or blocking MMP activity with TIMP-1, however, inhibits the formation of ectopic lesions.105 This model provides insight into a mechanistic link between steroidal regulation of MMP secretion in the establishment of an endometriosis-like disease and may prove useful in designing therapeutic agents to inhibit or minimize endometriosis lesions in the peritoneal cavity. It is still not understood, however, how misexpression of MMPs and TIMPs are due to an innate anomaly of the endometrium, immune system, or peritoneum of women with endometriosis. Mouse models of endometriosis have been recently reviewed,106 and the concept of resistance to progesterone action within endometriosis implants and in eutopic endometrium of women with and without disease are recent central themes that have increased our understanding of the pathogenesis of this disorder and suggest novel potential targeted therapies.
GROWTH FACTORS, ANGIOGENIC FACTORS, CHEMOATTRACTANTS, MORPHOGENS
Peritoneal fluid from women with endometriosis has mitogenic, angiogenic, morphogenic, and chemoattractant activities. It contains growth factors and cytokines secreted by monocytes/macrophages, as well as endometrial, ovarian, and mesothelial cells in the peritoneal cavity.107–109 These cytokines and growth factors can induce or suppress cell survival, proliferation, differentiation, angiogenesis, and the inflammatory response. In women with endometriosis, levels of proinflammatory cytokines are elevated, such as macrophage colony-stimulating factor,110 IL-1,111,112 lL-6113–115), and tumor necrosis factor-α (TNF-α),115–117 all of which are secreted by macrophages/monocytes. IL-1 and TNF-α, in turn, can stimulate the secretion of other cytokines such as IL-8,118,119 monocyte chemotactic protein-1,120,121 and RANTES (regulated upon activation, normal, T-cell expressed, and secreted),122,123 which are chemoattractants for neutrophils, T lymphocytes, monocytes, and macrophages, as well as a potent angiogenic factor (IL-8). These factors may contribute to pelvic pain115 or infertility.124
Growth factors may also play a fundamental role in the pathogenesis of endometriosis by independently stimulating cell survival, growth, or differentiation. Macrophage-conditioned medium contains mitogenic activity, some of which is attributable to TGF-β,125 platelet-derived growth factor (PDGF),115,126 and basic fibroblast growth factor (bFGF).127 Epidermal growth factor (EGF),127,128 insulin-like growth factors (IGFs),129–131 PDGF,115,126 and bFGF are potent mitogens for endometrial stromal cells in vitro. Hepatocyte growth factor (HGF) is a mitogen and morphogen for endometrial epithelial cells when co-cultured with stromal cells and may play a role in the regeneration of endometrial glands in ectopic locations.132 It is also an angiogenic factor. IGF-1 is an antiapoptotic growth factor and may enhance cell survival. EGF and IGF mediate estrogen actions in many tissues and thus are probably participants in the pathogenesis and pathophysiology of endometriosis.
Angiogenic activity is increased in peritoneal fluid from women with endometriosis.133 Vascular endothelial growth factor (VEGF), a potent angiogenic factor, has been found in peritoneal fluid from women with endometriosis, and VEGF levels in the peritoneal fluid correlate directly with the severity of disease.134 The source of VEGF has not been identified but may be the endometrial, mesothelial, or endothelial cells.
AROMATASE AND OTHER STEROIDOGENIC ENZYMES
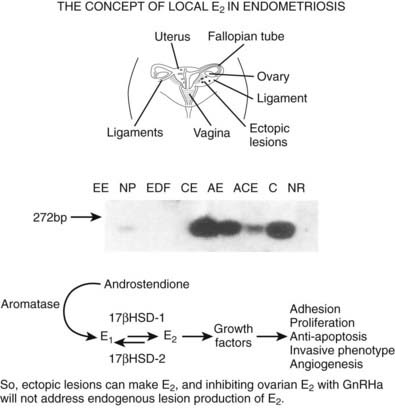
Recent evidence demonstrates abundant and abnormal expression in endometriotic lesions of aromatase, the key enzyme in estradiol (E2) biosynthesis.135 This enzyme is rarely expressed in eutopic endometrium of women without disease and is minimally expressed in women with disease.135 In addition, the enzyme that inactivates E2, 17β hydroxysteroid dehydrogenase-1, is down-regulated in endometriotic lesions,136 thus resulting in increased E2 synthesis locally in endometriotic lesions (Fig. 130-2) and contributing to the pathophysiology of progesterone resistance.137 This has important consequences with regard to therapies8,137 (see following section on treatment) which are aimed primarily at decreasing circulating E2 as a treatment strategy, insofar as local E2 production would not be treated.135
IMMUNE SYSTEM
There is ample evidence that endometriosis is associated with a variety of abnormal immune responses.138 Direct cytotoxic effects by peripheral and peritoneal natural killer (NK) cells in general and specifically against autologous endometrial cells in vitro are greatly reduced in women with endometriosis in comparison to controls.139–141 The NK cell activity, however, is partly under the regulation of soluble factors such as cytokines and growth factors, as evidenced by a further decrease in the activity of NK cells with the addition of sera or peritoneal fluid from women with endometriosis.141,142 In particular, NK cell activity is decreased,140,142,143 and endometrial cells are more resistant to NK-mediated cytolysis.140 Intercellular adhesion molecule-1 (ICAM-1) is important for NK–target cell interactions, including endometrial cell–NK cell interactions. Recent evidence suggests that endometrial cells from women with endometriosis produce more soluble ICAM-1,144 an antagonist of the NK-target interaction, which may contribute to the relative resistance of endometrial cell destruction by NK cells. These observations support the theory that women with endometriosis have a compromised ability to reject autologous endometrial tissue—for example, in the peritoneal cavity—and thus have an increased risk for the disorder. Whether this resistance to destruction by NK cells is an intrinsic immune dysfunction or a reaction to refluxed endometrial tissue that has been altered by the environment in the peritoneal cavity remains to be determined.143,145 Of interest, however, is the finding that ectopic endometrium could not be established in the peritoneum of NK-deficient mice.146
Alterations in immune function have been described in endometriosis, but the changes are complex, and none independently or satisfactorily explains the pathogenesis of this disorder. In addition, it is not clear whether the alteration is the cause of endometriosis or secondary to the inflammatory reaction that endometriosis induces. Further research involving the potential interactions between the immune system, MMPs, and TCDD may be promising.
ABERRANT GENE EXPRESSION
The use of rapidly emerging technologies that include high-throughput expression analysis by microarrays, proteomics, and accompanying strategies for data analysis (bioinformatics) have recently been applied to the study of endometriosis.147 Primarily two types of global gene expression profiling studies have been conducted on this tissue in humans: (1) transcriptome analysis of ectopic versus matched eutopic endometrium (whole tissue or specific cell types) and (2) transcriptome analysis of eutopic endometrium (whole tissue or specific cell types) from women with disease versus without disease.
ECTOPIC VERSUS EUTOPIC ENDOMETRIUM
In ovarian endometriomas, compared to paired eutopic endometrium during the proliferative phase of the cycle, genes regulating structural proteins and immune-related genes have been observed to be dysregulated.148 The significance of the up-regulation of genes associated with immune functions and structural proteins may reflect immune dysfunction contributing to the pathogenesis of the disorder and for anchoring lipid droplets for steroid-hormone biosynthesis by endometriosis tissue, respectively.148 A study using subtractive cDNA libraries and samples from subjects with minimal/mild peritoneal disease (revised American Fertility Society [rAFS] scores stage I/II) and moderate to severe peritoneal disease (stage III/IV) during either the proliferative or secretory phase revealed the following149: Several genes and gene families were identified, including extracellular matrix/cell adhesion proteins, ribosomal proteins, transcriptional regulators (including JUN and EGR1), RNA processing, signaling intermediates, cell-cycle regulators (CDK2), GDP/GTP binding proteins, metabolism, and other functions. Some of these genes are known to participate in estradiol action and have antiapoptotic actions, suggesting that they may play a role in the pathophysiology of the disorder and may be diagnostic biomarkers or candidate therapeutic targets.
Epithelial cells isolated by laser capture microscopy (LCM) from eutopic endometrium and ectopic endometrium (endometrioma and peritoneal lesions) and subsequent analysis using cDNA microarrays with 9600 genes revealed that samples cluster by site (ovarian endometrioma versus nonovarian disease), underscoring that ovarian and peritoneal endometriosis have different complements of gene expression and are likely different disorders.150 Both types of endometriosis had lower expression of genes involved in cell adhesion, Wnt signaling, and induction of apoptosis; both types had higher expression of genes involved in acute-phase response, cell proliferation, cell-cycle regulation, and regulation of transport. Differences were observed in expression of genes associated with glycoprotein function, response to oxidative stress, and G protein–coupled receptor signaling. This may represent a response to the proinflammatory environment in which the lesions exist. In fact, IL-8 was up-regulated, and IL-15 and PDGF-RA were down-regulated, consistent with other studies.151,152 Members of the MAPK pathway and oxidative stress pathways were up-regulated in endometriosis lesions compared to normal endometrium. The study by Wu et al.150 underscores the differences in gene expression in ovarian and peritoneal disease and some important pathways that have been implicated in the pathogenesis of endometriosis.34
Matsuzaki et al.152 characterized the transcriptome of deep endometriosis (i.e., rectovaginal disease) compared to ovarian and peritoneal disease using cDNA microarrays and LCM. PDGF-RA, PKCb1, and JAK1 were up-regulated, and sprouty 2 and MAP kinase kinase 7 were down-regulated in rectovaginal endometriosis stromal cells, supporting a role for the RAS/RAF/MAPK signaling pathway in the pathogenesis of the disorder. In endometriosis epithelial cells, COUP-TF2 and PGE2/EP3 were down-regulated, and since these are negative regulators of aromatase, their down-regulation may contribute to the known estradiol synthesis that occurs in some endometriotic lesions (see earlier). Tyrosine kinase receptor B (TRkB) in endometriosis epithelium and the serotonin transporter (5HTT) and the mu opioid receptor (MOR) in endometriosis stromal cells were up-regulated genes that may be candidates in the pathophysiology of pain in endometriosis.152
Using a cDNA microarray approach, Taylor and colleagues153 found differential responsiveness to IL-1β, an inflammatory cytokine, in endometrial stromal cells from eutopic endometrium and endometriomas. IL-1β down-regulated Tob-1 in endometriotic stromal cells but had minimal effect on normal stromal cells, suggesting that this cytokine promotes growth of endometriotic lesions through inhibition of Tob-1 and an association of IL-1β with altered cell-cycle gene expression in cells derived from endometriotic implants.153
Overall, these data highly suggest that ectopic and eutopic endometrium differ in their gene expression in subsets of endometriosis (e.g., peritoneal, deep lesions, and endometriomas) and demonstrate a dysregulation in ectopic endometrium of genes that are members of the Ras, MAP kinase, and PI3 kinase signaling pathways. This is consistent with the mouse model of overexpression of K-ras and conditional PTEN deletion in ovarian surface epithelium that resulted in endometriosis (and endometrioid ovarian carcinoma).34
Eutopic Endometrium
Global transcriptome analysis has also given insight into differences in eutopic endometrium of women with versus without endometriosis and molecular mechanisms underlying the pathogenesis of this disorder, as well as providing an opportunity to identify endometriosis biomarkers. Using differential display, Taylor and colleagues154 found up-regulation of a zinc-finger transcription factor coded by the early growth response (EGR)-1 gene in ectopic and eutopic endometrium of women with endometriosis compared to normal eutopic endometrium. EGR-1 is up-regulated by IL-1β, IL-6, TNF-α (inflammatory cytokines), and estradiol and may enhance angiogenesis via regulation of VEGF and its receptors. Upstream regulators of EGR-1 are associated with endometriosis, and this gene has been proposed to play a central role in the pathophysiology of endometriosis.
Using high-density oligonucleotide microarrays, Kao and colleagues151 performed global gene profiling of 12,686 genes during the window of implantation from eight subjects with and seven subjects without endometriosis. Dysregulation of a number of genes, including aromatase, progesterone receptor, and angiogenic factors, was determined as likely contributing to the pathophysiology of endometriosis. Interestingly, kallikrein was 100-fold up-regulated in the endometrium of women with endometriosis. Tissue kallikrein belongs to a family of serine proteases involved in the generation of bioactive peptide kinins in many organs. These enzymes may participate in proteolysis of extracellular matrix, which may be important in the establishment of the disease in the peritoneal cavity. Genes such as B61 may participate in the neovascularization and survival of endometrium, leading to the establishment of endometriosis. Of note also is the down-regulation of N-acetylglucosamine-6-O-sulfotransferase, glycodelin, IL-15, and Dickkopf-1, and up-regulation of semaphorin E. N-acetylglucosamine-6-O-sulfotransferase synthesizes a ligand for L-selectin important for blastocyst attachment to endometrial epithelium.155 Also, glycodelin and IL-15, progesterone-regulated genes in the peri-implantation period, are believed to play a major role in immunomodulation of the blastocyst implantation. These data suggest a poor response to progesterone in the endometrium during the implantation window in women with disease, which along with dysfunctions in embryonic attachment, survival, and signaling are mechanisms postulated to be operational in eutopic endometrium of women with endometriosis that may contribute to the pathogenesis of infertility associated with this disorder.
Recently, Burney et al.9 investigated the whole genome transcriptome of eutopic endometrium in the late proliferative and early and midsecretory phases in women with severe peritoneal disease (no ovarian endometriomas) compared to women without disease. Of note was the marked persistence of estrogen-regulated genes in early secretory endometrium (ESE) from women with disease at a time when progesterone action should inhibit these genes. In the proliferative-secretory transition, the fingerprint of persistence of cellular mitosis and minimal responsiveness of classically progesterone-regulated genes in ESE was also apparent in midsecretory endometrium (MSE). Principal component analysis revealed that ESE specimens from women with disease clustered closer to proliferative endometrium specimens than ESE from women without disease, confirming the observed resistance to progesterone action. Also, several genetic loci associated with endometriosis were identified among the dysregulated candidate genes. These data underscore progesterone resistance in endometrium of women with endometriosis and are consistent with inadequate clinical response to progestins in women with this disorder.151
With regard to gene expression in late secretory endometrium (LSE), Sherwin and colleagues recently reported a limited number of genes dysregulated during this period of progesterone withdrawal in women with versus without endometriosis.156 They concluded that this phase of the cycle was uninformative from a diagnostic perspective, whereas ESE was much more so as reported by Burney et al.9
Matsuzaki and colleagues157 recently determined cell-specific differences in gene expression in eutopic late PE, ESE, MSE, and LSE of women with and without deep endometriosis, using LCM and cDNA microarray analysis. They found up-regulation in LSE of uPAR in epithelial cells and KSR (a MAPK scaffold of the Ras pathway), and PI3K p85 regulatory subunit α in stromal cells. The involvement of two important signaling pathways, RAS/RAF/MAPK and PI3K, is strikingly similar to results with ectopic lesions of deep endometriosis compared to ovarian endometriomas and peritoneal disease (see earlier). The data overall suggest a common dysregulation of signaling in eutopic and ectopic endometrium in women with endometriosis among pathways that participate in cell-cycle regulation and cell survival, with the ectopic lesions being a more extreme phenotype.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


