Fig. 5.1
Hematoxylin and eosin micrograph of classic medulloblastoma. There are dense sheets of small round blue cells with scant cytoplasm
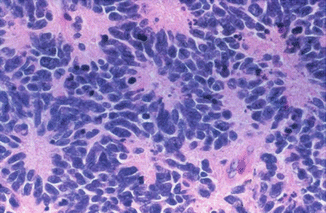
Fig. 5.2
Hematoxylin and eosin micrograph of Homer-Wright rosettes in medulloblastoma
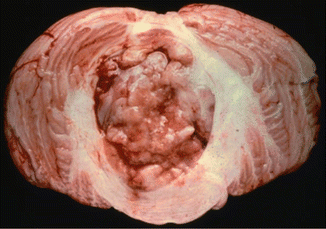
Fig. 5.3
Gross specimen of circumscribed medulloblastoma at the level of the cerebellar vermis and fourth ventricle
Microscopically, desmoplastic medulloblastoma is characterized by reticulin-rich stroma surrounding nodular foci of tumor in a so-called pale island configuration (Fig. 5.4). Tumors with these characteristics often arise at the superficial edge of a cerebellar hemisphere in adolescent or young adult patients where they cause a brisk inflammatory reaction of the meninges (Fig. 5.5) (Levy et al. 1997). In general, desmoplastic medulloblastoma has a more favorable outcome than other histopathologic variants, especially in infants, but this may be due in large part to co-occurrence with favorable molecular pathways (Kool et al. 2012; McManamy et al. 2007).
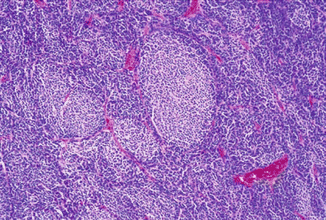
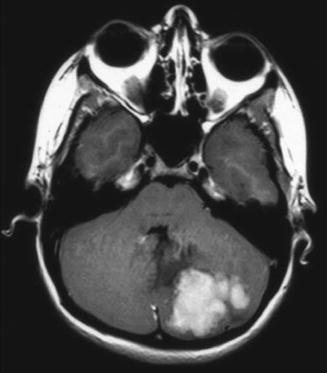
Fig. 5.4
Hematoxylin and eosin micrograph of desmoplastic medulloblastoma with pale islands of tumor cells and intervening dense reticulin
Fig. 5.5
Transverse T1-weighted axial magnetic resonance image following contrast administration demonstrating a superficial left cerebellar desmoplastic medulloblastoma in a 16-year-old girl
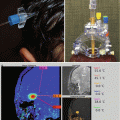
Large-cell medulloblastoma is identified histologically by pleomorphic, large round-to-irregular nuclei with prominent nucleoli, abundant cytoplasm, numerous mitoses, and a high apoptotic rate (Fig. 5.6) (Giangaspero et al. 1992). Synaptophysin and chromogranin expression are common, as are bulky spinal metastases (Fig. 5.7) and an aggressive disease course with 5-year overall survival as low as 10 % (Brown et al. 2000).
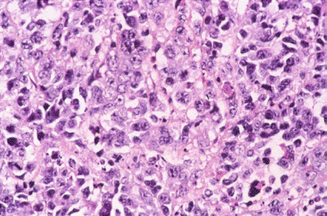
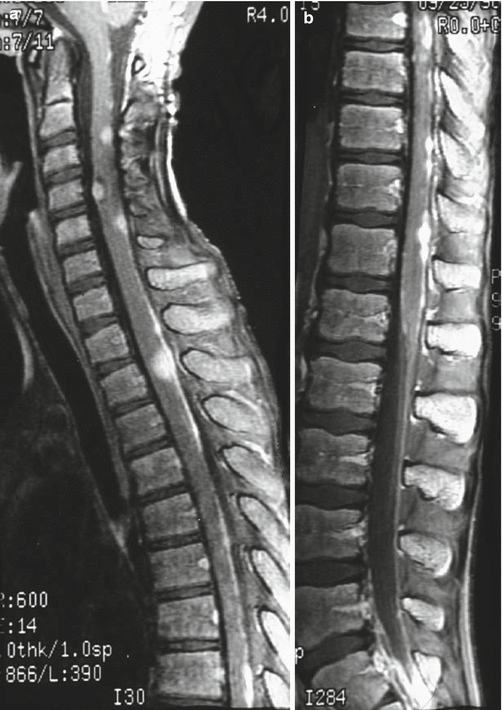
Fig. 5.6
Hematoxylin and eosin micrograph of large-cell medulloblastoma with cells having abundant cytoplasm, irregular nuclei, and prominent nucleoli
Fig. 5.7
T1-weighted sagittal post-gadolinium images of the thoracic (a) and lumbar spine (b) of a 10-year-old with widely metastatic large-cell medulloblastoma just after resection of a vermian primary mass. Widespread tumor dissemination is visualized on the surface of the cerebellum and spinal cord
Similar to large-cell tumors, anaplastic medulloblastoma is characterized by nuclear pleomorphism and molding, a high mitotic rate, and prominent apoptosis. Though these changes can be seen focally in all histopathologic subtypes of medulloblastoma, these histological findings are widespread throughout large-cell tumors. Since anaplastic regions are also common in large-cell tumors, many providers advocate for adoption of a unified large-cell/anaplastic category (McManamy et al. 2003).
Desmoplastic and nodular medulloblastomas, which are similarly sometimes unified under a joint desmoplastic/nodular designation, have significantly better outcomes than large-cell/anaplastic tumors. Other histopathologic variants are exceedingly rare, including medullomyoblastoma, characterized by striated muscle or muscle antigen expression, and melanotic medulloblastoma which is identified by melanin-forming neuroepithelial cells (Fowler and Simpson 1962; Marinesco and Goldstein 1933). Both these subtypes may carry a worse prognosis than classic medulloblastoma, but their respective incidences are too low to accurately calculate outcomes.
5.2.2.2 Molecular Features and Cytogenetics
Improved characterization of the histologic variants of medulloblastoma has also led to an improved understanding of the underlying biologic features. Early studies of cytogenetic and molecular features identified a variety of prognostic factors (Table 5.1). Proliferative index, as determined by Ki-67 antigen/MIB-1 staining, and the rate of apoptosis have both been found to influence outcome as well (Grotzer et al. 2001; Haslam et al. 1998).
Table 5.1
Cytogenetic and molecular features associated with medulloblastoma outcome
Good prognosis |
Hyperdiploidy (Gajjar et al. 1993) |
β-expression nuclear immunoreactivity (Ellison et al. 2005) |
Poor prognosis |
Isolated 17p loss of heterozygosity (Gilbertson et al. 2001) |
Overexpression of calbindin-D28k (Pelc et al. 2002) |
Cytogenetic studies have demonstrated that isochromosome 17q is present in approximately 50 % of cases, almost always in conjunction with loss of heterozygosity at 17p and most commonly in large-cell medulloblastoma (Bigner et al. 1988; Brown et al. 2000). Chromosome 1 rearrangements and 1q loss have been also variably noted in medulloblastoma (Bigner et al. 1988). In general, hyperdiploidy is associated with a better outcome than diploidy, but all cytogenetic features are prognostic only insofar as they affect molecular signaling pathways (Gajjar et al. 1993). In that regard, early investigations of molecular markers in medulloblastoma demonstrated associations with the loci containing c-myc, erbB2, β-catenin, and trkC (Ellison et al. 2005; Gajjar et al. 2004; Gilbertson et al. 2001; Grotzer et al. 2000; Kim et al. 1999). Whereas elevated expression of c-Myc and mutation of ErbB2 are both associated with unfavorable outcomes, high expression of the neurotrophic receptor gene trkC and β-catenin confers a more favorable prognosis. Amplification of the c-myc proto-oncogene is particularly associated with poor prognosis and perhaps unsurprisingly is frequently found in large-cell medulloblastomas associated with tumor anaplasia (Brown et al. 2000; Eberhart et al. 2002; Scheurlen et al. 1998).
Although genetic syndromes account for less than 5 % of all medulloblastoma diagnoses, molecular profiling of tumors from these patients provided an early understanding of the distinct molecular features that dominate sporadic and germline cases alike. For instance, germline mutations of the human homolog of the Drosophila gene Patched (PTCH), which has an inhibitory effect on the Sonic Hedgehog (Shh) pathway, are responsible for both medulloblastoma and basal-cell carcinoma in Gorlin syndrome patients, which is also known as nevoid basal-cell carcinoma syndrome (Raffel et al. 1997). Follow-up studies identified mutations in PTCH and other Shh pathway members such as GLI1 and SUFU in sporadic medulloblastoma cases as well (Pietsch et al. 1997; Raffel et al. 1997; Taylor et al. 2002; Wolter et al. 1997; Xie et al. 1997). Overexpression of PTCH and downstream Shh target genes, including GLI and N-myc, is highly correlated with desmoplastic medulloblastoma, and consistently, patients with these genomic aberrations are thought to have a relatively favorable clinical course (Pomeroy et al. 2002a). Transgenic mouse models of medulloblastoma further demonstrate that inactivation of the PTCH allele leads to medulloblastoma formation in a minority of animals, an incidence that is increased by concurrent TP53 mutation (Wetmore et al. 2000, 2001). Notably, patients with Li-Fraumeni syndrome, which is caused by loss of p53 function, are also at risk for medulloblastoma.
Turcot syndrome, which includes familial adenomatous polyposis (FAP), is defined by the simultaneous presence of brain tumors and inherited colonic polyposis. Medulloblastoma is found in association with mutation of the APC gene in FAP, which normally functions downstream of Wnt to assemble a protein complex that constitutively degrades β-catenin. In the absence of APC, β-catenin is trafficked to the nucleus to promote expression of diverse pro-growth genes. Somatic driver mutations are found in a subset of sporadic medulloblastoma patients and, much like cytogenetic abnormalities of the β-catenin locus, are generally associated with a more favorable outcome (Hamilton et al. 1995; Vasen et al. 2008; Zurawel et al. 1998).
5.2.2.3 Molecular Subgroups
Since 2006, high-throughput bioinformatic techniques such as genome-wide DNA copy number analysis, mRNA expression profiling, somatic copy number aberrations, whole-genome sequencing, and whole-exome sequencing have greatly enhanced our understanding of the molecular determinants of medulloblastoma in two ways (Cho et al. 2011; Hovestadt et al. 2014; Kool et al. 2012, 2008; Northcott et al. 2014, 2011a, b, 2012b; Pugh et al. 2012; Rausch et al. 2012; Robinson et al. 2012; Thompson et al. 2006). First, using unbiased sequencing techniques, these studies corroborate the prognostic significance of cytogenetic and molecular markers, many of which have a long history of clinical application. Second, the body of genomic evidence supports the existence of distinct molecular subgroups that dictate clinical outcomes. Although a variety of molecular subgroups have been proposed, the most widely accepted system separates medulloblastoma into Wnt, Shh, group 3 and group 4 tumors (Table 5.2) (Taylor et al. 2012). Among the various schema for clinical prognostication and stratification of patients with medulloblastoma – which include age, presence or absence of metastases, extent of resection, and histologic subtype – transcriptional profiling studies have demonstrated that molecular subgroup is the most important factor for risk assessment.
Table 5.2
Molecular subgroups of sporadic medulloblastoma
Wnt | Shh | Group 3 | Group 4 | |
|---|---|---|---|---|
Pediatric prevalence | ~10 % | ~30 % | ~25 % | ~35 % |
Peak incidence | Adolescents and young adults | Infants and young adults | Between infancy and adolescents | Adolescents |
5-year survival | ~95 % | ~60–80 % | ~40–50 % | ~75 % |
Cell of origin | Lower rhombic lip progenitors | GNPs from EGL and cochlear nuclei; SVZ stem cells | GNPs from EGL | Unknown |
Histologya | Classic | Nodular/desmoplastic | Classic and LC/A | Classic |
Genetic correlate | Turcot syndrome (FAP) | Gorlin syndrome (NBCCS) | Unknown | Unknown |
TP53 status | ~15 % rate of co-mutation, equivalent prognosis | ~20 % rate of co-mutation, worse overall survival (~40 %) | Uncommon | Rare in conjunction with isochromosome 17, but common with MYCN amplification; prognostic significance unclear |
Adult tumor characteristics | ~10–20 % incidence, worse prognosis | ~50–70 % incidence, comparable outcome | Extremely rare, worse prognosis | ~20–30 %, prognostic significance unclear |
Among the four principal molecular subgroups, Wnt and Shh tumors are named for the developmental signaling pathways that are thought to drive medulloblastoma formation. The molecular cornerstones of group 3 and group 4 tumors are less well characterized, although mutations in the Shh transcription factor GLI1 are common in both (Northcott et al. 2014). Indeed, the data suggest that subsets exist within each molecular subgroup, especially group 3 and group 4 neoplasms, and several high-quality review articles are available that thoroughly review these complexities (Batora et al. 2014; Kool et al. 2012; Northcott et al. 2012a).
Wnt subgroup tumors have a very low rate of metastatic spread and are associated with an excellent prognosis (Gajjar et al. 1993; Kool et al. 2012). Long-term survival for these patients exceeds 90 %, and as such, many individuals may live long enough to experience complications of therapy, rather than succumbing to recurrent disease. Wnt tumors account for approximately 10 % of sporadic cases, are equally distributed between males and females, and typically occur in adolescents and young adults (Ellison et al. 2005; Gajjar et al. 2004; Gilbertson et al. 2001; Grotzer et al. 2000; Kim et al. 1999; Northcott et al. 2012a, b). Although they may occur in the setting of any histopathologic variant, Wnt tumors generally exhibit classic histology (Brown et al. 2000; Eberhart et al. 2002; Gibson et al. 2010; Scheurlen et al. 1998).
Wnt peptides are a collection of highly conserved, lipid-modified glycoproteins which bind to the Frizzled family of transmembrane receptors to transduce intercellular signals upstream of APC and β-catenin. Although first identified as a proto-oncogene in a mouse model of breast cancer, autocrine and paracrine Wnt signaling are critical for embryonic development through regulation of cell migration and proliferation, body axis patterning, and cell fate determination (Nusse and Varmus 2012; Raffel et al. 1997). Deregulation of the canonical Wnt pathway is associated with numerous malignancies, including skin, breast, lung, esophageal, colorectal, prostate, ovarian, and brain cancer (Anastas and Moon 2013; Pietsch et al. 1997; Raffel et al. 1997; Taylor et al. 2002; Wolter et al. 1997; Xie et al. 1997). With respect to the pathogenesis of medulloblastoma, alterations in Wnt signaling that promote stabilization and nuclear localization of β-catenin are common to both syndromic and sporadic tumors. In particular, somatic mutations of CTNNB1, the gene that encodes β-catenin, and loss of chromosome 6 are present in the majority of sporadic Wnt medulloblastomas. As such, β-catenin immunohistochemistry has become a routine component of pathological analysis following resection of medulloblastoma.
Hedgehog proteins, such as Shh, are secreted glycolipoproteins that control cell fate and proliferation by binding to the transmembrane protein Ptch. In the presence of Shh, Ptch-mediated repression of Smoothened (Smo) is released. In turn, Smo triggers a signal transduction pathway that culminates in activation of Gli transcription factors (Corbit et al. 2005; Pomeroy et al. 2002; Stone et al. 1999). Shh pathway-associated medulloblastoma accounts for approximately 30 % of all sporadic cases, is slightly more common in males, and occurs primarily in infants and adults (Al-Halabi et al. 2011; Archer et al. 2012; Kool et al. 2012; Wetmore et al. 2000; 2001). These tumors are rarely metastatic upon diagnosis, are frequently associated with nodular/desmoplastic histology, and are considered to have an intermediate prognosis with approximate overall survival ranging from 60 % to 80 % (Archer et al. 2012; Hamilton et al. 1995; Vasen et al. 2008; Zurawel et al. 1998). Given the prevalence of this molecular signature, immunohistochemistry for GAB1, a transmembrane mediator of Shh signaling, is routinely performed on all medulloblastoma resection specimens.
Large-cell anaplastic medulloblastoma, which has an aggressive clinical course, is most commonly associated with the molecular subtype known as group 3 (Kool et al. 2008; Leonard et al. 2001; Northcott et al. 2011; Thompson et al. 2006). Rarely found in adults or infants, and associated with a mere 40–50 % overall survival, nearly half of these tumors present with metastatic spread at the time of diagnosis (Kool et al. 2012). Males are approximately twofold more likely than females to be effected with group 3 medulloblastoma, which accounts for approximately 25 % of all cases.
In general, group 3 medulloblastoma is characterized by high levels of Myc amplification and genomic instability including gain of chromosomes 1q, 7, and isochromosome 17q, as well as loss of chromosomes 10q, 11, 16q, and 17p (Cho et al. 2011; Northcott et al. 2011). Group 4 medulloblastomas, which comprise 35 % of all cases, are also characterized by cytogenetic abnormalities, including isochromosome 17q and heterozygous loss of the X chromosome, in addition to amplification of CDK6 and N-Myc (Kool et al. 2012). Usually found in adolescent males and associated with classic histology, approximately 40 % of group 4 medulloblastomas are associated with metastases at diagnosis. Less is known about the biology of group 3 and group 4 tumors, although both are unified by specific collections of epigenetic and structural DNA variations, as opposed to misregulation of a canonical developmental pathway such as Wnt or Shh. Mechanistic investigations of group 3 and group 4 medulloblastomas are therefore required to better understand the pathophysiology of these tumors and to identify targets for molecular therapies. In that regard, the Shh antagonist vismodegib has been trialed for medulloblastoma, but responses are near-universally transient. Indeed, subsequent mechanistic and genomic studies have shown that medulloblastoma cells are capable of rapid evolution to develop resistance to single-agent molecular therapy (He et al. 2014; Rudin et al. 2009; Kool et al. 2014; Yauch et al. 2009). As such, combination therapy may be necessary if molecular agents are to be successfully incorporated into the treatment regimen for medulloblastoma.
5.2.3 Clinical Features
Children with medulloblastoma frequently present with signs and symptoms of obstructive hydrocephalus and cerebellar dysfunction over the course of 2–6 months due to obstruction of the fourth ventricle. Early symptoms may include irritability, behavioral changes, and declining school performance. Infants are frequently found to display macrocephaly, splitting of the cranial sutures, or a bulging anterior fontanel. Many patients progress to develop emesis, particularly upon awakening, horizontal diplopia, head tilt, clumsiness, ataxia, and headaches. Seizures may occur, as can symptoms associated with leptomeningeal dissemination, including cranial nerve dysfunction, pituitary abnormalities such as precocious puberty, and distal neurologic deficits from spinal metastases. Late neurologic signs include strabismus, ataxia, or weakness (Honig and Charney 1982). Untreated, the symptoms from mass effect and obstructive hydrocephalus invariably lead to progressive neurologic deterioration and death. Every effort should therefore be made for expedient relief of CSF pressure and an oncologic resection of tumor, as discussed below.
5.2.4 Natural History
At diagnosis, 14–43 % of medulloblastoma patients are found to have microscopic or nodular seeding in the subarachnoid space of the spine or brain (Deutsch and Reigel 1980; Tarbell et al. 2000). Indeed, medulloblastoma has the greatest tendency for subarachnoid space seeding and extraneural spread among all pediatric brain tumors. In less than 5 % of cases, tumors spread outside the CNS (Kleinman et al. 1981).
Medulloblastoma relapse most often occurs at the primary tumor site or elsewhere within the cerebellum, with or without neuraxis spread (Halberg et al. 1991). Median time to recurrence is 14 months from diagnosis, although for infants, the time to progression is just 6 months (Duffner et al. 1993; Minn et al. 2001). Subclinical subarachnoid tumor spread is not uncommon at autopsy, although these data are derived from children treated with 36 Gy of craniospinal irradiation (CSI), or infants treated without radiotherapy. In total, approximately 75 % of patients present with neuraxial dissemination at the time of recurrence, but it is not known how the pattern and timing of relapse will change with variation of prophylactic neuraxial radiation dose, different systemic and intrathecal chemotherapy regimens, or molecular targeted agents.
Children diagnosed before 3 years of age have significantly worse survival than older patients, but there do not appear to be significant differences among age groups of 4–9 years, 10–14 years, and 15–19 years (McNeil et al. 2002). Outcome is independent of race (McNeil et al. 2002). It is debated whether the generally adverse effect of very young age on outcome may be confounded by the frequent absence of irradiation during treatment of these patients. However, emerging evidence suggests a high incidence of molecular mutations with a particularly aggressive natural history that may explain the trend toward impaired survival in this patient population (Kool et al. 2012).
5.2.5 Diagnostic Imaging
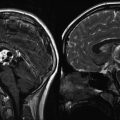
Although imaging has not historically played a role in the definitive diagnosis of medulloblastoma, central nervous system imaging is universally employed for children presenting with the clinical features described above. Most often for the sake of expediency, as well as due to the frequent need for pediatric anesthesia during magnetic resonance imaging (MRI), patients will initially undergo a computed tomography (CT) scan of the head. A high-quality CT scan will almost always detect a posterior fossa mass. Classically, medulloblastoma appears as a cerebellar mass that is hyperattenuated due to its high cellular density (Fig. 5.8). These lesions homogeneously enhance following contrast injection, and approximately 20 % of lesions contain intratumoral calcifications (Bourgouin et al. 1992; Poretti et al. 2012). Nonetheless, anatomic definition and preoperative planning require a high-quality MRI scan. Children with large posterior fossa masses detected on CT or MRI are at high risk for neurologic deterioration from transient increases in intracranial pressure (ICP) while supine or sedated during a prolonged MRI screening. However, there is risk of shunt failure due to retained blood products in the ventricles after resection of tumor, and placement of a shunt increases the risks of intratumoral hemorrhage and upward herniation. For these reasons, shunting is not advocated before resection, and most patients are managed with steroids. External ventriculostomy may be considered in emergency scenarios, and in general, shunt placement does not appear to increase the risk of neuraxial or systemic tumor spread (Berger et al. 1991).
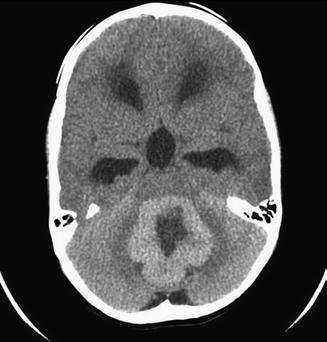
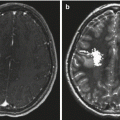
Fig. 5.8
Axial noncontrast CT scan of a patient with a hyperdense medulloblastoma
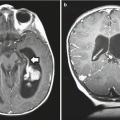
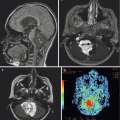
The appearance of medulloblastoma on MRI can be varied. On T1-weighted images, the mass can be either homogenous or heterogeneous in consistency, is generally iso- or hypointense, and avidly enhances with contrast administration (Fig. 5.9). Large-cell/anaplastic tumors in particular may demonstrate ring enhancement as a result of tumor necrosis. T2-weighted images have similarly been reported to vary with histologic subtype: classic tumors may display T2 signal hyperintensity from cysts and calcifications, and desmoplastic/nodular lesions may be isointense with internal heterogeneity on T2 sequences. If possible, a full spine MRI should be obtained preoperatively to determine if there is leptomeningeal dissemination, the presence of which markedly affects long-term outcome. At the most extreme end of this spectrum, a minority of patients present with diffuse leptomeningeal dissemination which is described as “sugar coating” of the brain and spine on post-contrast T1-weighted MR images (Fig. 5.10). If an MRI of the spine cannot be obtain preoperatively, it is recommended that 10–14 days be allowed to pass after surgery to allow for reabsorption of blood products that collect within the thecal sack following craniotomy and may obscure evaluation for tumor nodules. In contrast, it is imperative that the postoperative restaging scans of the posterior fossa are completed within 24–72 h of resection before granulation tissue, gliosis, and blood products obscure residual tumor.
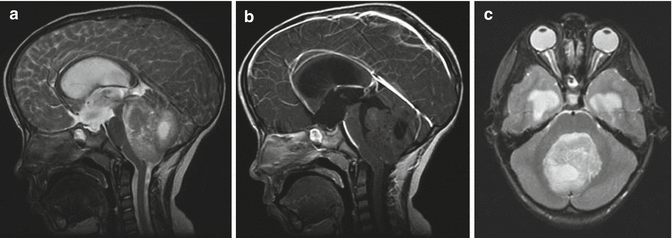
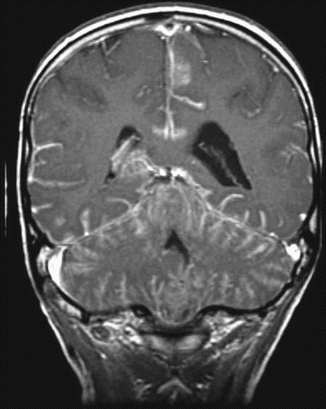
Fig. 5.9
(a–c) Magnetic resonance images of a 5-year-old boy with a typical posterior fossa medulloblastoma. (a) T2-weighted sagittal image showing a large heterogeneous partially cystic mass arising within the cerebellar vermis, displacing the brainstem anteriorly and with associated ventricular enlargement. (b) T1-weighted image with contrast demonstrates minimal enhancement, which is a common finding. (c) T2-weighted axial image showing that the tumor extends to the floor of the fourth ventricle but does not invade the brainstem
Fig. 5.10
A coronal T1-weighted magnetic resonance image following contrast administration. Disseminated leptomeningeal tumor is clearly seen as a bright layer of tissue “sugar coating” the brain
5.2.6 Treatment
5.2.6.1 Surgery
Virtually all children with a posterior fossa mass undergo craniotomy, the goals of which are to relieve mass effect, obtain a tissue diagnosis, reestablish CSF circulation, and cytoreduction to facilitate adjuvant therapy. In general, there is no indication for stereotactic or open biopsy, unless the cerebellar tumor is diffuse or there is extensive leptomeningeal seeding. Even in such instances, symptomatic relief following primary tumor debulking is more durable than after CSF diversion alone and certainly lessens the risk of death from mass effect and brainstem compression. In cases without evidence of metastases, every effort should be made for maximal safe resection. Although children with less than 1.5 cm2 of residual disease on postoperative imaging have an improved prognosis for long-term, relapse-free survival, these data were largely obtained during the CT era, and it is unclear how well they generalize to MRI studies (Zeltzer et al. 1999a). As brainstem infiltration does not affect overall prognosis, removal of tiny tumor foci in this region or lying at the floor or exit of the fourth ventricle is not warranted due to the significant risk of morbidity and mortality associated with these surgical procedures (Duffner et al. 1993; Zeltzer et al. 1999b).
Following primary tumor resection, external ventriculostomies are usually weaned in an intensive care setting over the first week or 10 days by either gradually elevating or clamping the external drain. If ICP rises with weaning of the ventriculostomy, the presence of hydrocephalus must be presumed and a permanent ventriculoperitoneal shunt should be placed. Similarly, if the ventriculostomy can be removed successfully, ventricular size and clinical symptoms must continue to be observed carefully as delayed hydrocephalus can still occur in approximately 30 % of patients.
5.2.6.2 Staging
Following resection and histopathological diagnosis of medulloblastoma, a lumbar puncture for CSF cytology should be obtained 10–14 days postoperatively to assess for microscopic tumor spread. Ventricular sampling of CSF for cytology has inferior sensitivity and should not be used, unless a lumbar sample absolutely cannot be obtained (Gajjar et al. 1999). Although bone-marrow biopsy is not recommended, a bone or positron emission tomography scan may be obtained if there is evidence of disseminated disease.
The classic system of medulloblastoma staging separates patients into two risk groups based upon patient age, extent of resection, and presence or absence of metastases (Tables 5.3 and 5.4) (Chang et al. 1969b). Notably, primary tumor size is not prognostic and does not factor into this system. Average risk includes children greater than 3 years old, less than 1.5 cm2 residual tumor, and no evidence of either gross or microscopic metastases. High risk is defined by more than 1.5 cm2 residual, patient age less than 3 years, and M+ disease. Notably, the prognostic significance of micrometastatic disease has been debated, as two trials have demonstrated no difference in the overall survival of patients with M1 and M0 medulloblastoma. Moreover, these staging criteria have been challenged due to the omission of histopathologic data and tumor molecular subtype. Indeed, ongoing clinical trials are beginning to incorporate these features to stratify patients, and it is likely that the formal staging system for medulloblastoma will be amended to incorporate biologic features in the future.
M0 | No evidence of gross subarachnoid or hematogenous metastasis |
M1 | Microscopic tumor cells within the cerebrospinal fluid |
M2 | Gross nodular seeding in the cerebellar, cerebral subarachnoid space, or in the third or lateral ventricles |
M3 | Gross nodular seeding in spinal subarachnoid space |
M4 | Extraneural metastasis |
Table 5.4
Risk stratification for medulloblastoma
Average risk |
<1.5 cm2 postoperative residual tumor, stage M0, and age >3 years |
High risk |
>1.5 cm2 postoperative residual tumor, stage M1-4, or age ≤3 years |
5.2.6.3 Radiotherapy
Adjuvant treatment for medulloblastoma most often commences with radiotherapy beginning approximately 3–4 weeks after surgery because of time required for wound healing, neurologic recovery, and staging. Since most patients requiring CSI are children, special considerations are often necessary, such as proper stabilization during treatment and assistive anesthesia. In addition, many children have neurologic deficits impairing their ability to lie motionless for any length of time.
Baseline assessments of endocrine and cognitive function should be performed during the postoperative period as CSI may adversely affect the pituitary and thyroid glands along with the cerebrum. Numerous studies have quantified the long-term sequelae of CSI in pediatric patients, which also include skeletal deformity from stunted vertebral body growth. Along with endocrine and neurocognitive dysfunction, these toxicities are intrinsically related to patient age at the time of treatment, as well as the dose of radiation administered (Mabbott et al. 2005; Merchant et al. 2009; Palmer et al. 2013). Processing speed and memory seem to be most affected, although the combined effects of craniotomy and chemotherapy certainly contribute to all of these findings.
At present, a radiation dose of 23.4 Gy to the craniospinal axis plus a boost to 54 Gy to the posterior fossa followed by chemotherapy is the standard of care for average-risk disease (Packer et al. 2006). This approach has resulted in 5-year overall survival of approximately 85 %, but given the aforementioned treatment-associated toxicities, reduced-dose craniospinal irradiation of 18 Gy and conformal radiotherapy boost to the tumor bed are active areas on ongoing clinical investigation. Given their uniquely favorable prognosis, WNT-driven average-risk medulloblastoma patients constitute a particularly appealing cohort for which to diminish therapy intensification, including reduced CSI dose to 18 Gy to limited target volume boost to a total of 54 Gy. Intensification of chemotherapy with autologous peripheral blood stem-cell support has also been reported and may offer one potential avenue for reducing the dose of ionizing radiation to the craniospinal axis (Gajjar et al. 2006a). In high-risk disease, 36 Gy to the craniospinal axis plus a boost at the posterior fossa to 55.8 Gy, followed by chemotherapy, is the standard therapy and results in 5-year survival rates of 50–70 %.
The CSI field arrangement for medulloblastoma radiotherapy classically consists of opposed lateral brain portals and a posterior spinal axis field, all of which are matched in the region of the cervical spine with the patient in the prone position. The posterior fossa is typically treated with at least 1 cm margin, and the inferior border is set at C2. However, treatment of the entire posterior fossa also delivers a significant dose to the parietal, occipital, and temporal lobes, as well as the parotid glands, cochlea, pharynx, and other associated structures. Therefore, ongoing cooperative studies are evaluating whether the target volume reduction to the primary site tumor bed alone in average-risk medulloblastoma can be performed without compromising disease control. Indeed, many pediatric oncologists have already incorporated this approach into practice.
Beyond reducing the total dose of CSI and the boost target volume, alternative radiation modalities such as proton-beam and helical intensity-modulated radiotherapy (IMRT) (e.g., TomoTherapy®) are increasingly in use in an effort to mitigate potential long-term side effects of radiation. Outcome data for each are limited, and benefits are largely theoretical. However, comparisons of proton beam, conventional 3D radiation, and IMRT for treatment of the posterior fossa and spinal column suggest superior sparing of normal structures by protons, likely with mitigation of long-term toxicities (St Clair et al. 2004).
5.2.6.4 Chemotherapy
Attempts to omit CSI in children less than 3 years or exclusion of the entire neuraxis from the radiation field have resulted in reduced survival (Bouffet et al. 1992). However, the morbid sequela of CSI, along with the poor survival of children with high-risk medulloblastoma, has led to sustained efforts by cooperative groups to introduce chemotherapy and reduce or delay radiation. Indeed, these efforts have at least allowed for radiation to be reserved for salvage in select infants (Ashley et al. 2008; Duffner et al. 1993; Geyer et al. 2005; Grill et al. 2005; Rutkowski et al. 2005).
Chemotherapy was formally introduced for medulloblastoma in the late 1970s, when the International Society of Pediatric Oncology (SIOP I), the Children’s Cancer Study Group (CCG 942), and the Pediatric Oncology Group (POG 7909) each performed prospective randomized trials of CSI alone versus postradiation chemotherapy. These studies investigated different systemic cytotoxic regimens, but were alike in the heavy application of DNA alkylating agents such as lomustine and procarbazine, as well as the microtubule poison vincristine (Evans et al. 1990; Krischer et al. 1991; Tait et al. 1990). Although chemotherapy did not improve overall survival for all children, a benefit was apparent in patients with bulky residual or metastatic disease. In sum, these findings and a series of subsequent trials demonstrated that medulloblastoma is one of the most chemotherapy-sensitive of all brain tumors. Alkylators and platinum compounds are the foundation of adjuvant chemotherapy, particularly lomustine and cisplatin, and sometimes procarbazine, cyclophosphamide, ifosfamide, or carboplatin. Vincristine is often administered weekly during radiation and then again during adjuvant chemotherapy. In addition, the topoisomerase II inhibitor etoposide is active in disseminated and infant forms of the disease (Ashley et al. 1996; Duffner et al. 1993). Finally, methotrexate, an antimetabolite, has been used in European trials, but is uncommon in the United States due to an association with radiation-induced leukoencephalopathy.
In average-risk medulloblastoma, a series of trials from SIOP, the French Society of Pediatric Oncology (SFOP), the Children’s Oncology Group (COG, the result of a merger between POG and CCG), and the German Society of Pediatric Oncology (GPO and HIT) have attempted to reduce CSI by adding either preradiation neoadjuvant chemotherapy or, more commonly, postradiation chemotherapy (Table 5.5) (Bailey et al. 1995; Gajjar et al. 2006a; Gentet et al. 1995; Kortmann et al. 2000; Kuhl et al. 1998; Packer et al. 2006, 1999; Taylor et al. 2001; Thomas et al. 2000). POG 8631/CCG 923 treated patients without chemotherapy and either 23.4 Gy or 36 Gy CSI, but was suspended in 1990 when an interim statistical analysis revealed an elevated rate of relapse in the reduced-dosage radiotherapy group (Thomas et al. 2000). Follow-up over time demonstrated that patients treated with 36 Gy experienced an event-free survival of 67 % at 5 years. Event-free survival was marginally inferior in the 23.4 Gy group, at 52 % after 5 years (p = 0.077), but there was also an increased rate of early relapse and isolated exoprimary recurrence, contrary to earlier limited institutional experiences (Deutsch et al. 1996; Halberg et al. 1991). Subsequently, CCG 9892 employed 23.4 Gy CSI plus a boost to the posterior fossa to a total of 55.8 Gy with concurrent weekly vincristine, followed by eight cycles of lomustine, cisplatin, and vincristine. By this approach, 5-year event-free survival from average-risk disease reached 78 % and was statistically no different than the POG 8631/CCG 923 benchmark using 36 Gy. While CCG 9892 accrued just 65 eligible patients, the results suggested that chemotherapy could be substituted for at least a portion of CSI. In support of this hypothesis, COG A9961 randomized average-risk medulloblastoma patients to receive either lomustine or cyclophosphamide in combination with cisplatin and vincristine following 23.4 Gy CSI and a whole posterior fossa boost to 54 Gy. Both arms achieved >80 % event-free survival at 5 years (Packer et al. 2006).
Table 5.5
Cooperative group studies for average-risk medulloblastoma, age ≥3 years
Study | Years of accrual | Number of eligible patients | Preirradiation chemotherapy × cycles | Craniospinal radiotherapya | Postirradiation chemotherapy × cycles | Percent event-free survival at 5 years | Comments |
|---|---|---|---|---|---|---|---|
SIOP IIb,c (Bailey et al. 1995) | 1984–1989 | 40 | None | 35 Gy | None | 60 ± 8 69 ± 8 75 ± 7 42 ± 8 | No significant benefit from “sandwich” chemotherapy, but negative interaction between “sandwich” chemotherapy and reduced irradiation |
36 | None | 25 Gy | None | ||||
38 | PCZ/VCR/MTX × 1 | 35 Gy | None | ||||
36 | PCZ/VCR/MTX × 1 | 25 Gy | None | ||||
SFOP M7 b,d (Gentet et al. 1995) | 1985–1988 | 31 | ″1-ln-11e × 2,HD | 30–37.5 Gyf | None | 74 | |
MTX × 2 | |||||||
POG 8631/CCG 923b (Thomas et al. 2000) | 1986–1990 | 44 | None | 36 Gy | None | 67 ± 7 52 ± 11 | Increased incidence of early, exoprimary neuraxis relapse |
44 | None | 23.4 Gy | None
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|