Abstract
This chapter describes the causes, clinical manifestatilons, diagnosis, and treatment of various quantitative disorders of white blood cells, both inherited and acquired. It also discusses disorders of white cell function, diagnostic work-ups, and treatment.
Keywords
Leukocytosis, neutropenia, eosinophilia, lymphocytosis, leukocyte function disorders
White blood cells (WBCs) are cells of the immune system that defend the body against infectious disease and foreign material. The total WBC and the differential counts are valuable guides in the diagnosis, treatment, and prognosis of various childhood illnesses. The normal leukocyte counts and the absolute counts of different classes of leukocytes vary with age in children and their ranges are listed in Appendix 1 .
It is important to calculate the absolute count of each class of WBC rather than relative percentage count for the purposes of quantitative interpretation. If nucleated red blood cells (NRBCs) are present, the total WBC count includes the total nucleated cell count (TNCC). Under these circumstances, the true total WBC count is calculated by subtracting the absolute NRBC count from the TNCC. This correction is generally required in the hemolytic anemias and in newborns when NRBCs tend to be raised.
Blood smear examination of the white cell morphology is important in the diagnosis of various causes of leukocytosis. For example, in severe infections or other toxic states, the neutrophils may contain fine deeply basophilic granules (toxic granulations) or larger basophilic cytoplasmic masses (Döhle bodies). Vacuolization of neutrophils may also occur. Döhle bodies are also found on examination of the blood smear in pregnancy, burns, cancer, leukemia blasts, May Hegglin anomaly, and many other conditions.
Quantitative Disorders of Leukocytes
Leukocytosis
Leukocytosis is an increase in the total WBC count more than two standard deviations above the mean for age. It is most commonly due to an increase in the absolute number of mature neutrophils (neutrophilia), but can be due to an increase in the absolute numbers of lymphocytes, monocytes, eosinophils, or basophils. Leukocytosis may be acute or chronic. Table 13.1 lists the causes of leukocytosis and Table 13.2 lists the causes of neutrophilia.
|
|
|
In infants and children, there is a tendency to release immature granulocytes into the circulation, and the WBC count may reach very high levels (>50,000/mm 3 ). This is called a leukemoid reaction and is often associated with bacterial infections. The term left shift is a >5% increase in the percentage of immature precursors (primarily bands) due to rapid release of the bone marrow reserve. The shift to the left may be so marked as to suggest myeloid leukemia. Table 13.3 lists the distinguishing features of leukemoid reaction and true leukemia.
| Feature | Leukemoid reaction | Leukemia |
|---|---|---|
| Clinical | Evidence of infection | Hepatosplenomegaly |
| Lymphadenopathy | ||
| Hematologic | No anemia | Anemia |
| No thrombocytopenia | Thrombocytopenia | |
| Bone marrow | Normal, hypercellular | Blasts |
| Decreased megakaryocytes | ||
| Decreased erythroid precursors | ||
| Leukocyte alkaline phosphatase | High | Absent |
Monocytosis is defined as an absolute monocyte count of >500–800/mm 3 . Table 13.4 lists the causes of monocytosis and monocytopenia. The causes of basophilia, defined as an absolute basophil count >200/mm 3 are listed in Table 13.5 . Eosinophils and lymphocytes are discussed later in this chapter.
|
|
Leukopenias
Leukopenia exists when the total WBC count is less than 4000/mm 3 . Leukopenia may result from a decrease in one or more specific classes of leukocyte. The causes of neutropenia are listed in Table 13.6 and lymphopenia in Table 13.7 . Leukopenia can result from a number of conditions. However, isolated leukopenia resulting from a decrease in all classes of leukocytes is observed uncommonly.
|
|
a A case associated with Coxsackievirus B2 has been described.
Neutropenia
Neutropenia is defined as a decrease in the absolute neutrophil count (ANC). The ANC is calculated by multiplying the total WBC count by the percentage of segmented neutrophils and bands: ANC=WBC (cells/mm 3 )×percent (segmented+bands). In Caucasians, neutropenia is defined as an ANC of less than 1000/mm 3 in infants between 2 weeks and 1 year of age and less than 1500/mm 3 beyond 1 year of age. African Americans may have lower counts, with ANC levels 200–600/mm 3 less than in Caucasians. Neutropenia can be transient or chronic. Neutropenia is considered “chronic” when it persists beyond 3 months and is due to reduced production or increased destruction of neutrophils. It may be an inherited, intrinsic disorder or an acquired, extrinsic defect.
In pseudoneutropenia, a normal neutrophil population may be shifted toward the marginating compartment, leaving fewer cells in the circulating compartment. The WBC count measures only the circulating cells and not the marginating pool; therefore, this represents a pseudoneutropenia. The bone marrow is normal in appearance. The neutrophils function normally and the leukocyte changes are usually found incidentally on blood count. Marginating neutrophils may be uncovered by the injection of epinephrine.
The severity and duration of neutropenia correlate with susceptibility to develop various types of bacterial infections. The severity of neutropenia is graded according to ANC as follows:
- •
Mild neutropenia: ANC 1000–1500/mm 3
- •
Moderate neutropenia: ANC 500–1000/mm 3
- •
Severe neutropenia: ANC less than 500/mm 3
Clinical Manifestations
Patients with severe neutropenia are at an increased risk of infection from their own endogenous bacterial flora that reside in the mouth, oropharynx, gastrointestinal tract, and skin. For this reason, the frequency of Gram-negative bacterial infections and Staphylococcus aureus infections is high in these patients. The risk of infection is inversely proportional to the ANC. With mild neutropenia, stomatitis, gingivitis, and cellulitis may develop. More severe infections occur when the ANC is below 500/mm 3 , with perirectal abscesses, colitis, pneumonia, and sepsis being common. Neutropenia alone does not predispose to parasitic, viral, or fungal infections unless other parts of the immune system are compromised, such as postchemotherapy.
Figure 13.1 shows an approach to the diagnosis of neutropenia.
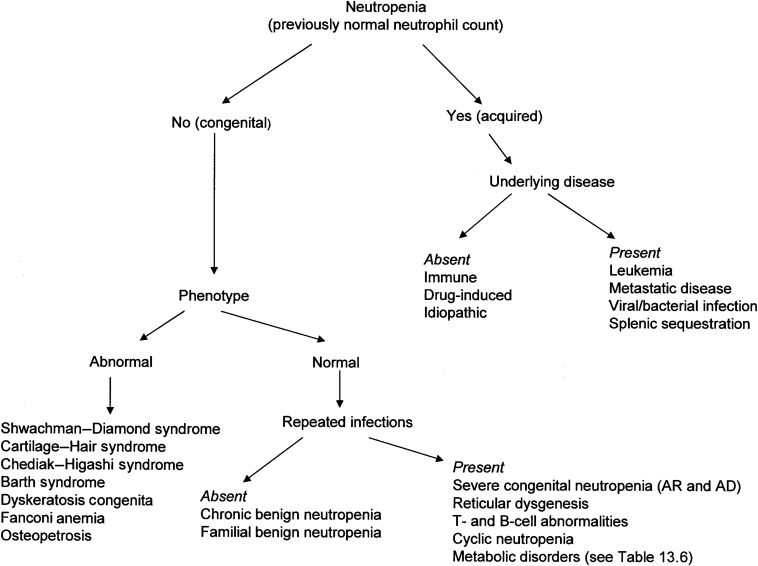
Decreased Production or Intrinsic Defects of Neutrophils
The inherited congenital neutropenias are primary bone marrow failure disorders involving the myeloid series. These are discussed in more detail in Chapter 8 .
Table 13.6 lists the congenital neutropenias.
Benign Ethnic Neutropenia
Benign ethnic neutropenia is observed in a variety of populations, including Africans, West Indians, Yemenite and Ethiopian Jews, Bedouin Arabs, and Jordanians. In these groups, an ANC as low as 1000/mm 3 may be considered as normal. There is no increased infection risk in these individuals.
Neutropenia Associated with X-linked Agammaglobulinemia
This primary humoral immunodeficiency syndrome is inherited as an X-linked recessive trait. It is caused by mutations in the gene encoding a tyrosine kinase known as Bruton’s or B-cell tyrosine kinase (BTK). BTK is expressed in all stages of B-cells lineage development, and in myeloid maturation.
Clinical Manifestations
- •
Severe hypogammaglobulinemia, antibody deficiencies, and increased susceptibility to infection.
- •
Most patients present by 5 years of age.
- •
15–25% of patients may develop neutropenia, which can be severe, during the stress of active infections when rapid production of these cells is required. Those with neutropenia are more prone to develop fungal or Pneumocystis jirovecii infections.
- •
Laboratory findings include severe decrease in serum IgG, IgA, and IgM levels and marked decrease in B-lymphocytes, but normal T-cell function.
Treatment
- •
Immunoglobulin therapy and aggressive antibiotic regimens for infections.
- •
Short-term use of Granulocyte Colony Stimulating Factor (G-CSF) has been found to be effective in some reported cases, but no evidence-based data are available.
Neutropenia Associated with Autosomal Recessive Agammaglobulinemia
There are at least six forms of autosomal recessive agammaglobulinemia. Mutations of the gene encoding for the μ heavy chain, surrogate light chain, and Ig-alpha are found in 85–90% of patients affected by this disease. Similar clinically to X-linked agammaglobulinemia (XLA), there are absent B-cells and associated neutropenia.
Treatment
Immunoglobulin replacement therapy, antibiotics, and a short course of G-CSF may be used, although no evidence-based data are available.
Neutropenia Associated with Abnormal Cellular Immunity in Cartilage Hair Hypoplasia
Cartilage hair hypoplasia is a rare autosomal recessive disorder found more commonly in Amish and Finnish populations. It is due to mutations in the ribonuclease mitochondrial RNA-processing gene that result in clinical heterogeneity.
Clinical Manifestations
- •
Short-limbed dwarfism, fine hair, moderate to severe neutropenia, and impaired cell-mediated immunity. There is a greater susceptibility to infection in children, including sino pulmonary, and a higher incidence of leukemia/lymphoma among adults.
- •
Hematologic findings: Lymphopenia, macrocytic anemia, neutropenia, immune-mediated thrombocytopenia (ITP), autoimmune hemolytic anemia (AIHA).
Treatment
- •
Allogeneic stem cell transplantation corrects both the immunologic defects and neutropenia. G-CSF therapy has been effective in one case report.
Neutropenia Associated with Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) is a heterogeneous group of disorders involving immune dysfunction of B- and T-lymphocytes.
Clinical Manifestations
- •
Defined as an age-specific reduction in serum IgG. IgM and IgA levels vary.
- •
Presence of B-cells with poor or absent response to immunizations, and absence of other immunodeficiency states.
- •
Usual onset is between 20–30 years of age and only occasionally in childhood. Most patients manifest recurrent infections of the sinopulmonary and GI tracts. They are also prone to develop noncaseating granulomas of the lung, skin, gut, and other organs.
- •
These patients are predisposed to develop autoimmune disorders such as ITP, systemic lupus erythematosus (SLE), AIHA, rheumatoid arthritis, and thyroiditis.
Treatment
Neutropenia in CVID is explained on an autoimmune basis. Patients respond to G-CSF treatment, IV immunoglobulin, and antibiotics.
Myelokathexis and WHIM Syndromes
WHIM is a rare primary immunodeficiency disorder associated with warts, hypogammaglobulinemia, recurring bacterial infections, and retention of neutrophils in the bone marrow. If warts are absent, the condition is called “myelokathexis.”
Genetics
The majority of patients have an autosomal dominant mutation of CXCR4 that prevents the normal release of mature neutrophils from the marrow into the blood.
Clinical Manifestations
- •
Moderate to severe neutropenia.
- •
WBC count is usually <1000/mm 3 with severe neutropenia and lymphocytopenia. Neutrophils and eosinophils contain vacuoles, prominent granules, and nuclear hypersegmentation with pyknotic nuclei connected to each other by thin filaments. Normal morphology of lymphocytes, monocytes, and basophils.
- •
Neutrophil function is usually normal.
- •
Bone marrow shows granulocytic hyperplasia and neutrophils with similar degenerative changes as in the blood.
Treatment
- •
Immune globulin replacement therapy can reduce bacterial infections.
- •
G-CSF or granulocyte–macrophage colony-stimulating factor (GM-CSF) results in increased neutrophils.
- •
Preliminary evidence suggests that long-term treatment with the CXCR4 antagonist, plerixafor, is effective in increasing circulating leukocytes and decreasing infection frequency.
Selective IgA Deficiency and Neutropenia
Selective IgA deficiency is the most common form of immunodeficiency. Infections most commonly affect the respiratory and GI tracts. Neutropenia may be autoimmune in nature. There are no reports of the efficacy of G-CSF in this condition.
Dubowitz Syndrome
Dubowitz syndrome is an autosomal recessive disorder characterized by dysmorphic facies, mental retardation, microcephaly, growth retardation, eczema, and recurrent neutropenia.
Laboratory findings include low IgG and IgA with elevated IgM levels.
Neutropenia Associated with Hyperimmunoglobulin M Syndrome
The hyperimmunoglobulin M syndromes are rare heterogeneous disorders in which defective immunoglobulin (Ig) class switch recombination leads to normal or increased levels of serum IgM associated with deficiency of IgG, IgA, IgE and poor antibody function. These can be genetic or acquired secondary to congenital rubella syndrome, phenytoin use, or T-cell malignancy.
Genetics
- •
The most common form is an X-linked recessive trait, caused by mutations in the gene for CD40 ligand ( TNFSF5 ). It has associated abnormal cellular immunity and patients develop infections by opportunistic organisms including P. jirovecii , histoplasmosis, Cryptosporidium , and toxoplasmosis.
- •
Two rare hyper-IgM syndromes with autosomal recessive inheritance and normal T-cell function are caused by defects in the gene for enzymes called activation-induced cytidine deaminase and uracil nucleoside glycosylate. These are humoral immunodeficiencies.
Clinical Manifestations
- •
Presents in infancy with severe recurrent bacterial infections due to encapsulated organisms or opportunistic organisms including P. jirovecii , histoplasmosis, Cryptosporidium , and Toxoplasma .
- •
Neutropenia is common. It may be transient, cyclic (10%), or chronic (50% of cases). Mechanism is not known but antineutrophil antibodies are not detected.
- •
DAT (direct antiglobulin test—Coombs)-positive AIHA and thrombocytopenia may be present.
- •
Lymphoid hyperplasia and predisposition to lymphoid malignancy.
- •
Low serum IgA, IgE and IgG; elevated IgM.
- •
Number of circulating B-cells is normal or increased and T-cells are normal.
Treatment
- •
Immunoglobulin replacement therapy reduces bacterial infections.
- •
Neutropenia responds to G-CSF.
- •
Hematopoietic stem cell transplantation is reported to be curative in patients with CD40L mutations.
Neutropenia Associated with Metabolic Diseases
The presenting clinical features in neutropenia associated with metabolic diseases ( Table 13.6 ) are lethargy, vomiting, ketosis, and dehydration during the neonatal period, failure to thrive, and growth retardation. The marrow is hypoplastic in these conditions with decreased numbers of myeloid precursors. Idiopathic hyperglycinemia and methylmalonic acidemia also have associated thrombocytopenia. Individuals with isovaleric acidemia have a characteristic odor of “smelly feet.”
Glycogen Storage Disease Type 1b
- •
Glycogen storage disease type Ib (GSD-1b) is a rare autosomal recessive disorder characterized by hypoglycemia, hepatosplenomegaly, seizures, and failure to thrive in infants.
- •
It results from a deficiency of glucose-6-phosphate-translocase enzyme and is characterized by impaired glucose homeostasis.
Clinical Manifestations
- •
Patients with GSD-1b frequently have neutropenia and/or neutrophil dysfunction and are susceptible to recurrent bacterial infections involving the perirectal area, ears, skin, and urinary tract. Life-threatening infections occur less frequently.
- •
Mechanism of neutropenia in GSD-1b may be caused by both increased levels of apoptosis and egress of neutrophils from the blood to the tissues.
Treatment
- •
G-CSF corrects neutropenia, defects in neutrophil chemotaxis, and intracellular bacterial killing, reducing the incidence of infections.
Barth Syndrome
- •
Rare X-linked recessive disorder characterized by cardiomyopathy, mild neutropenia, proximal skeletal myopathy, growth retardation, mitochondrial abnormalities, neutropenia, and increased urinary excretion of 3-methylglutaconic acid.
- •
Due to mutations in the TAZ gene (G4.5) resulting in an inborn error of lipid metabolism.
- •
Neutropenia is variable (absent to severe; persistent, intermittent, or cyclical). Despite the mild neutropenia, their cardiac defect places them at increased risk for morbidity when they develop infections.
Treatment
- •
G-CSF is used concomitant with appropriate prophylactic antibiotics if clinically indicated. Long-term G-CSF may be used based on severity.
Bone Marrow Disease
Bone marrow failure from any cause may present with neutropenia as a component of the pancytopenia that occurs in these disorders. These include:
- •
Congenital (e.g., Fanconi anemia, dyskeratosis congenita) aplastic anemias.
- •
Acquired (idiopathic or secondary) aplastic anemias.
- •
Bone marrow infiltrative disorders (non-neoplastic storage diseases such as Gaucher disease, Niemann–Pick disease).
- •
Neoplastic (leukemia, neuroblastoma).
- •
Osteopetrosis.
Bone marrow failure syndromes are discussed in detail in Chapter 8 .
Increased Destruction or Extrinsic Defects of Neutrophils
Drug-Induced Neutropenia
Drug-induced neutropenia is defined as an idiosyncratic reaction to an offending drug. Onset may be days to months after exposure and may be acute or insidious. It may be due to:
- •
Idiosyncratic suppression of myeloid production affecting a few exposed persons—for example, antibiotics (novobiocin, methicillin), sulfonamides, antidiabetics (tolbutamide, chlorpropamide), antithyroids (propylthiouracil, methimazole), antihistamines, and antihypertensives (chlorothiazides, Aldomet).
- •
Regularly occurring dose-dependent myeloid suppression from cytotoxic drugs or antimetabolites—for example, 6-mercaptopurine, methotrexate, and nitrogen mustard.
- •
Toxic suppression of myeloid production in the marrow due to differences in individual ability to metabolize a drug—for example, phenothiazine and thiouracil.
- •
Drug-hapten disease, in which antibodies to the drug–leukocyte complex are produced. Clinically presents with rash, fever, lymphadenopathy, hepatitis, or pneumonia—for example, amidopyrine-related drugs (dipyrone, phenylbutazone), sulfapyridine, phenobarbital, mercurial diuretics, and chlorpropamide.
Treatment
- •
Immediate withdrawal of the offending drug is indicated.
- •
G-CSF may be indicated for persistent neutropenia or if severe infections develop.
Immune Neutropenia
Neonatal Immune Neutropenia—Alloimmune and Autoimmune
Immune neutropenias may be alloimmune (isoimmune), due to alloantibodies directed against epitopes (most commonly involving HNA-1a, HNA-1b, and HNA-1c) inherited from the father, analogous to Rh isoimmunization. In infants born to mothers with autoimmune neutropenia, antibodies are due to transplacental transfer of IgG antibodies against both maternal and fetal cells (e.g., a mother who has systemic lupus erythematosus).
Clinical Manifestations
Neutropenia may be moderate to severe. Infants may be asymptomatic or they may develop sepsis, cellulitis, omphalitis, or pneumonia.
- •
Neutropenia usually resolves by 2 months of age but may last up to 6 months.
- •
Bone marrow is hypercellular with an increase in neutrophil precursors and a paucity of mature neutrophils.
Treatment
- •
Antibiotics for the treatment and prevention of infections.
- •
Immunoglobulin or G-CSF therapy have been effective for more serious infections.
Autoimmune Neutropenia
Autoimmune neutropenia can be primary or secondary.
Primary Autoimmune Neutropenia
Chronic autoimmune neutropenia of childhood is a common disorder resulting from antibodies against leukocytes. It is analogous to AIHA or autoimmune thrombocytopenia. Neutrophil antibodies may adversely affect the function of neutrophils, producing qualitative defects in the neutrophils and amplifying the risk of infection associated with neutropenia. Neutrophil autoantibodies may also affect myeloid precursor cells. When this occurs, it can produce profound neutropenia. The disease is characterized by the following:
- •
Age of onset: 3–30 months; median age, 8 months.
- •
Neutropenia may be mild to severe. During infections, the ANC may transiently increase to normal levels.
- •
Physical examination is normal with mild splenomegaly noted occasionally.
- •
Benign infections of skin and upper respiratory tract, as well as otitis media, are seen in 90% of cases. These are not usually life-threatening and tend to be responsive to standard antibiotics.
Diagnosis
- •
Neutrophil counts range from 0 to 1000 cells/mm 3 . Monocytosis is common.
- •
The bone marrow may be normal or may show evidence of myeloid hyperplasia with marked reduction in segmented neutrophils due to their destruction by antibodies.
- •
Epinephrine or hydrocortisone administration results in a rise in neutrophil count.
- •
Antineutrophil antibodies are not always detectable and screening has to be repeated for antibody detection from a reliable reference laboratory. Immunoassay is more sensitive than leukoagglutination testing for diagnosing immune neutropenia. In most patients, the autoantibody is auto-anti-HNA1 and some have auto-anti-HNA2.
The different types of antineutrophil assays are:
- •
Granulocyte immunofluorescence test (GIFT) detects neutrophil-bound IgG by binding of glutaraldehyde-fixed patient neutrophils to fluorescent-labeled antihuman IgG.
- •
Granulocyte indirect immunofluorescence test (GIIFT) uses the patient’s serum with normal neutrophils or previously typed neutrophils and subsequent incubation with fluorescent-labeled antihuman IgG.
- •
Granulocyte agglutination test (GAT) uses the patient’s serum incubated with normal neutrophils followed by microscopic evaluation for leukoagglutination.
- •
Enzyme-linked immunoassay (ELISA) uses microtiter plates with bound glutaraldehyde fixed normal neutrophils to detect antineutrophil antibodies in patient’s sera. An antihuman IgG conjugated to a reporter enzyme is used for detection.
- •
Monoclonal antibody-specific immobilization of granulocyte antigens (MAIGA) is the most specific. It involves incubation of type-specific neutrophils with both patient’s serum and mouse monoclonal antibodies directed against another neutrophil surface antigen. The mixture is passed over an affinity column containing antimouse IgG antibodies and then is assayed for the presence of human IgG. This allows for the detection of antibody and knowing its specificity in one assay.
GIFT and GAT are used most commonly to diagnose autoimmune neutropenia.
Prognosis
Spontaneous recovery usually occurs within a few months to a few years. The median age at recovery is 30 months (range, 7–73 months). In 95% of cases, recovery occurs by 4 years of age.
Treatment
Infections are not related to the degree of neutropenia and respond to standard antibiotics. Usually infections are not life-threatening but in patients with severe neutropenia who develop severe or recurrent infections, the following management is recommended:
- •
Appropriate antibiotics for acute bacterial infections or prophylactic antibiotics such as trimethoprim/sulfamethoxazole.
- •
Mouth care with oral rinses and good dental hygiene for mouth ulcers or gingivitis.
- •
In patients with more serious infections, G-CSF can be used at low doses of 1–2 µg/kg to improve ANC>1000/mm 3 . The dose can be further decreased or given on alternate days or less often.
- •
Immunoglobulin therapy has been used with variable response and duration.
Secondary Autoimmune Neutropenia
Secondary neutropenia is seen in other acquired disorders of the immune system. It occurs more commonly in adolescents and adults than in children. In these patients, chronic immune neutropenia warrants screening for associated immunodeficiency or autoimmune disorders.
Autoantibody specificity: Pan-FcR γ IIIb.
The following diseases are associated with secondary autoimmune neutropenia:
- •
Evans syndrome.
- •
AIHA.
- •
Autoimmune thrombocytopenia.
- •
Thyroiditis.
- •
Insulin-dependent diabetes mellitus.
- •
Common variable immune deficiency.
- •
Systemic lupus erythematosus.
- •
Rheumatoid arthritis.
Treatment
- •
Treat the underlying condition.
- •
For patients with severe infection, low-dose G-CSF can be used.
Autoimmune Lymphoproliferative Syndrome
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of defects in lymphocyte apoptosis. ALPS is reviewed in detail in Chapter 16 .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


