Introduction
The thyroid axis is probably the best example of the physiological interactions between the mother, the fetus, and their environment. The discovery of the crucial role of maternal iodine intake for the normal development of the fetus also led to the first successful intervention in preventive medicine. Two centuries ago, the idea to use iodine to treat endemic goiter, which predominantly affected women, encountered a lot of resistance because it was believed that iodine was toxic for humans. The demonstration that 10% of the insoluble fraction of the thyroid was iodine and that it could be used to treat myxedema and goiter was followed by the first systematic clinical trial to assess the efficacy of iodized salt in preventing goiter. Iodized salt at the population level was next introduced and resulted in a marked decrease in the prevalence of goiter in children and, even more importantly, in the disappearance of “endemic cretinism.” However, pregnant women require double the iodine intake of nonpregnant women to maintain normal thyroid hormone concentrations, so they and their fetuses are most vulnerable to decreases in iodine intake at the population level. The iodine deficiency that resurfaces in adolescent girls in certain geographic areas is therefore worrisome and illustrates the need for continuous surveillance of intake of this essential nutrient. With this caveat, endemic goiter and cretinism have all but disappeared in the industrialized world and the most severe cases of congenital hypothyroidism (CH) are caused by developmental defects of the thyroid and, albeit much less frequently, to defective pituitary control of thyroid function. These are the focus of this chapter.
Embryology, physiology, and physiopathology
Development of the Thyrotrophic Axis
The Pituitary
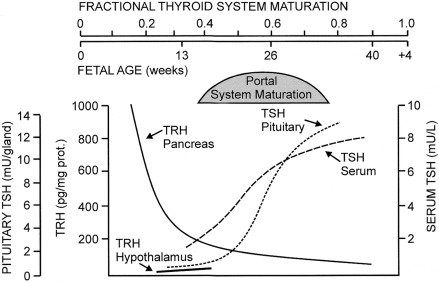
As for all organs, anatomic development of the hypothalamic-pituitary-thyroid system occurs during the first trimester of gestation. By 3 weeks of gestation, a series of homeodomain proteins or transcription factors begin to drive the differentiation of the human embryonic forebrain and hypothalamus. Immunoreactive thyrotropin-releasing hormone (TRH) becomes detectable in human embryonic hypothalamic by 8 to 9 weeks of postconceptional age and is also produced by the fetal gut and pancreas.
Anatomically, the pituitary gland develops from two ectodermal anlagen: a neural component from the floor of the primitive forebrain, and Rathke’s pouch from the primitive oral cavity. The latter is visible by 5 weeks, evolving to a morphologically mature pituitary gland by 14 to 15 weeks. The pituitary-portal blood vessels are present by this time and mature further through 30 to 35 weeks. A wide spectrum of congenital malformations collectively named midline defects , including holoprosencephaly and septooptic dysplasia, may be associated with central hypothyroidism and other anterior pituitary deficiencies. The molecular mechanisms underlying these malformations have been identified in some cases. Within the pituitary itself, PROP-1 and PIT-1 are terminal factors in the differentiation cascade of pituitary cells and PIT-1 or PROP-1 deficiency results in profound defects in growth hormone, prolactin, and thyroid-stimulating hormone (TSH) secretion, as well as age-dependent pituitary hypoplasia.
The Thyroid
The human thyroid gland also develops from two endodermal anlagen: the median anlage derives from the primitive pharyngeal floor and the two paired lateral anlagen from the fourth pharyngeal pouches. A branching morphogenesis program is essential for the thyroid to attain a normal size. The long-held belief that the lateral anlagen were the only source of calcitonin-producing cells has been challenged by the observation that sublingual thyroids, which are derived exclusively from the median anlage, contain calcitonin messenger ribonucleic acid (mRNA) and protein. Thyroid follicular cells can differentiate within the lateral anlagen, as illustrated by histological observations, as well as by patients in whom the only thyroid tissue is a lateral ectopy. Both the median and lateral structures are visible by day 16 to 17 of gestation; by 50 days, they have fused and the thyroid gland has migrated to its definitive location in the anterior neck. The thyroglossal duct, from the foramen cecum to the final location of the thyroid, may persist and is constituted of degenerated thyroid follicular cells. Within the thyroid gland, iodine concentration, TSH receptors, thyroglobulin (TG), and thyroperoxidase (TPO) mRNA and protein can be demonstrated by 70 days.
Thyroid embryogenesis depends on the expression of a programmed sequence of transcription factors, including thyroid transcription factors-1 and -2 (TTF-1 or TiTF-1, now designated as NKX2 homeobox 1 -NKX2.1-; and TTF-2, now Forkhead box E1 -FOXE1-) and paired box gene 8 (PAX8). In newborn mice, biallelic inactivation of Nkx2.1 results in the absence of both pituitary and thyroid glands, with complete absence of both thyroid follicular cells and of calcitonin-producing C-cells, whereas that of Pax8 results in a small thyroid gland composed almost exclusively of C-cells. FoxE-1 null mouse embryos have either an absent thyroid or an ectopic sublingual gland, but all newborn pups have athyreosis in addition to cleft palate. Mutations in the homologous genes, however, account for at most 2% of cases of thyroid dysgenesis in humans. Therefore genes extrinsic to the thyroid may be involved in the control of its migration.
Interactions Between Cardiovascular and Thyroid Development
The coordination of the development of the brain and of its vasculature suggests that brain and vessels may follow the same extrinsic cues and/or that there are reciprocal interactions between the two. By analogy, the lack of epithelial to mesenchymal transition during thyroid migration suggests it is not an active process but rather involves movement of the surrounding tissues and vessels. Further evidence that the development of the heart and vessels influences thyroid migration stems from the observation that a patient with cooccurring sublingual thyroid and congenital heart disease had a deletion of NETRIN1 and that ntn1a zebrafish morphants had abnormal thyroid morphogenesis, resulting from a lack of proper guidance by the dysplastic vasculature.
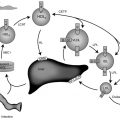
Maternofetal Transfers and Their Clinical Consequences ( Fig. 8.1 )
Iodine is an essential component of thyroid hormones. In this chapter, the term iodine will refer to both iodine itself (I 2 ) and iodide (I – ). The human placenta expresses the sodium–iodine symporter throughout gestation, which explains why the mother’s iodine status is reflected in the fetus. If the mother’s iodine intake is suboptimal, the fetal thyroid cannot constitute appropriate stores of iodine and fetal hypothyroidism will ensue. Worldwide, inadequate maternal iodine intake leading to late consequences remains a major public health problem, although an increasing number of emerging countries appear to have achieved iodine sufficiency. Prevention of intellectual disability in the offspring by supplying the mother with adequate iodine is a public health intervention with undisputed benefits.
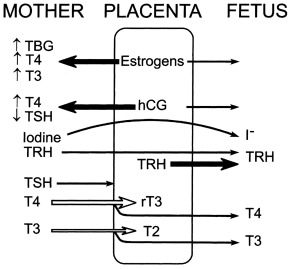
In contrast with iodine, thyroxine (T4) was for a long time thought not to cross the placenta in substantial amounts. However, several findings clearly indicate that maternal T4 must cross the placenta in physiologically relevant amounts. First, T4 is detectable in human embryonic tissues before the onset of fetal thyroid function and must therefore be of maternal origin. Second, the transfer of T4 from mother to fetus must continue later in gestation because the concentration in cord blood from neonates with complete absence of thyroid function is 30% to 50% of that of normal neonates. Third, birth weight and neonatal TSH levels are lower in infants born to a mother who has chronically higher T4 concentrations because of a mutation inactivating the thyroid hormone receptor who have not inherited the maternal mutation than in those who have. These data indicate that maternal T4 crosses the placenta in physiologically relevant amounts throughout gestation and that, in excess, has a direct toxic effect on the fetus.
However, low T4 concentrations in pregnant women do not appear to be causally related to a lower IQ in the children: two randomized controlled trials have shown a similar developmental outcome in the offspring of T4-treated versus placebo-treated hypothyroxinemic pregnant women. Moreover, in two case series of women with severe hypothyroidism diagnosed during pregnancy, but corrected by the third trimester, the intellectual outcome of the offspring was normal. Thus universal screening for thyroid dysfunction in the mother during pregnancy remains hotly debated. On the other hand, women with known hypothyroidism require closer monitoring during pregnancy, as 85% of women who are already receiving T4 therapy require a 30% to 50% increase in dose during pregnancy, because of the estradiol-induced increase in serum T4-binding globulin.
The transplacental transfer of T4 is not always sufficient to prevent the development of fetal goiter if the fetus has severe thyroid dyshormonogenesis ( Fig. 8.2 ). Fetal goiters may be large enough to interfere with the flow of amniotic fluid into the oropharynx, causing progressive hydramnios and eventual lung hypoplasia. In such cases, levothyroxine injected into the amniotic fluid is swallowed by the fetus, leading to a decrease in the size of the fetal thyroid and in the degree of hydramnios. The injection of thyroxine into the umbilical vein, which carries an even higher risk of triggering premature labor or fetal loss than amniocentesis, should be restricted to fetuses with a goiter that continues to increase in spite of repeated intraamniotic injections. Invasive and potentially risky procedures should not be undertaken to protect the brain of affected fetuses: indeed, the fetal brain is, to a large extent, protected from the deleterious effect of hypothyroidism through upregulation of brain type 2 deiodinase, which converts the prohormone T4 into its biologically active derivative, triiodothyronine (T3). This likely accounts for the observation that even in CH with delayed bone maturation at diagnosis (indicating a prenatal onset), the intellectual outcome is within normal limits if continuous and adequate treatment is instituted shortly after birth. Thus the in utero treatment of fetal hypothyroidism should only be considered for goiter causing progressive hydramnios. Although the identification of a goiter by prenatal ultrasound may be increasing, it remains rare and even direct examination at birth often fails to detect goiters that are obvious on imaging. Goiters can also be detected in fetuses borne by women with Graves disease (see section on congenital hyperthyroidism).

Although a pro-TRH molecule is produced by the placenta, TRH concentrations in the maternal circulation are very low and thus have little effect on fetal thyroid function. However, TRH, a tripeptide, crosses the placenta readily and, when injected into the mother, increases thyroid hormone concentrations in the fetus. Because thyroid hormones stimulate fetal lung maturation, maternal TRH treatment to decrease neonatal respiratory distress syndrome has been attempted, but with negative results.
Immunoglobulin(Ig)s of the IgG type cross the placenta, hence transient fetal/neonatal hyperthyroidism from transplacental transfer of TSH-receptor activating antibodies can occur in women with past or present Graves disease. On the other hand, when pregnant women are overtreated with antithyroid drugs, which also cross the placenta readily, their fetus may develop hypothyroidism; however, only one case of CH out of about 30,000 births is attributable to this in the Québec database (unpublished observations). In addition, the antithyroid medication has a short half-life relative to that of immunoglobulin, so that a baby with apparently normal thyroid function tests at birth may develop hyperthyroidism within several days of birth, as the effects of antithyroid medication wanes, allowing the effects of thyroid-stimulating immunoglobulin (TSI) to become apparent. Lastly, transient neonatal hypothyroidism from maternofetal transfer of TSH-receptor blocking antibodies can also occur but only accounts for 2% of cases of neonatal hypothyroidism identified by screening, and a specific screening strategy is not required for babies born to women with Hashimoto thyroiditis.
Thyroid function tests are often ordered clinically in newborns whose mothers have a history of thyroid disease and, if abnormal, require special consideration of optimal approach to therapy. For example, hypothyroidism at birth caused by treatment of maternal Graves disease with antithyroid medication may only require observation, in the expectation that the effects of the drugs will dissipate over a few days; hyperthyroidism may follow, albeit exceptionally. For hypothyroidism or hyperthyroidism resulting from maternal blocking or stimulating antibodies, respectively, treatment will be required as these effects may last several months.
Maturation of Thyroid Hormone Synthesis and Secretion ( Figs. 8.3 and 8.4 )
Maturation of thyroid function in the fetus reflects changes at the level of the hypothalamus, pituitary, and thyroid. Serum TRH concentrations are relatively high in the human fetus, because it is produced at both hypothalamic and extrahypothalamic sites and because the TRH-degrading activity of fetal blood is low. Fetal serum TSH increases from a low level at 18 to 20 weeks to a peak value of approximately 7 to 10 mU/L at term. After delivery, in response to exposure to the relatively colder extrauterine environment, there is an acute release of TSH with mean serum levels peaking at 30 minutes at concentrations of approximately 70 mU/L. A rapid decline follows within 24 hours, and levels return more gradually to 5 to 10 IU/L within the first week of life. The increase in serum T4 levels immediately after birth is TSH dependent. T3 rises as a result of both TSH-stimulated thyroidal production and peripheral conversion of T4 to T3. For newborn screening purposes, the percentage of samples with TSH greater than 15 mU/L, which is 9% in the first 24 hours, drops to 0.3%, as early as the second day, so any sample obtained after 24 hours can be used.
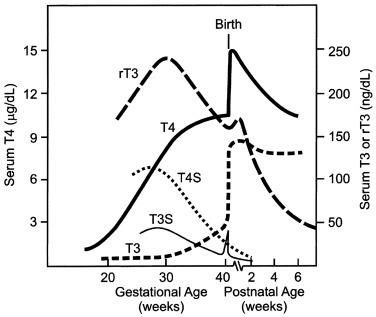
Only free thyroid hormones enter cells, and hormones bound to serum thyroxine-binding globulin (TBG) and other transport proteins are not available to tissues. In addition, T4 is a prohormone and it is T3 that is biologically active within the cell, so that deiodination of T4 is essential for tissue euthyroidism. Both serum transport proteins and intracellular deiodination change during development. As previously mentioned, the fetal thyroid gland is capable of iodine concentration and iodothyronine synthesis as early as 70 days of gestation, a reflection of a sharp increase in the expression of the sodium-iodine symporter and of the appearance of a follicular architecture. Starting at 18 to 20 weeks, TBG and total T4 concentrations in fetal serum increase steadily until the final weeks of pregnancy.
The study of free T4 (fT4) concentrations in fetal/neonatal blood has been hampered by the relative inadequacy of the commercially available immunoassay systems for measurements in these samples. The fetal serum T3 concentration remains low until 30 weeks because of two factors: first, the low activities of type I iodothyronine monodeiodinase result in relatively low rates of T4 to T3 conversion in fetal tissues; second, type III monodeiodinase in placenta and selected fetal tissues degrades T3 to T2. After 30 weeks, serum T3 increases slowly until birth. This prenatal increase in serum T3 is caused by progressive maturation of liver type I deiodinase activity increasing hepatic conversion of T4 to T3, and to decreased placental T3 degradation. Postnatally, T3 and T4 serum concentrations increase 2- to 6-fold within the first few hours, peaking on the second day of life. These levels then gradually decline to levels characteristic of infancy over the first 4 to 5 weeks of life. The deiodinase enzymes are described in greater detail later.
In the human, the fetal thyroid gland grows and its production increases under the influence of the increasing serum TSH level during the second half of gestation, as evidenced by the severely atrophic and hypofunctional glands observed in newborns with biallelic mutations that inactivate either the beta-subunit of TSH or the TSH receptor. On the other hand, the maturation of the negative feedback control system appears to occur earlier than previously thought because an elevated TSH in serum obtained at cordocentesis can be seen in fetuses with primary hypothyroidism as early as 18 weeks. Thyroid function in the premature infant is characterized by low circulating concentrations of T4 and fT4, a normal or low concentration of TSH, and a normal or prolonged TSH response to TRH, suggesting a degree of relative hypothalamic (tertiary) hypothyroidism (see Fig. 8.3 ).
In summary, fetal thyroid hormone secretion results from increasing hypothalamic TRH secretion and increasing thyroid follicular cell sensitivity to TSH and is regulated by increasing pituitary sensitivity to thyroid hormone inhibition of TSH release. The marked cold-stimulated TRH-TSH surge at birth is followed by a marked increase in T4 secretion and fT4 concentration with a new equilibrium reached by 1 to 2 months. During infancy and childhood, there is a progressive decrease in T4 secretion rate (based on a micrograms/kg per day) correlating with a decreasing metabolic rate.
Maturation of Thyroid Hormone Metabolism ( Fig. 8.5 )
The thyroid gland is the sole source of T4. Most of the circulating T3 after birth is derived from conversion of T4 to T3 via monodeiodination in peripheral tissues. Deiodination of the iodothyronines is the major route of metabolism, and monodeiodination may occur either at the outer (phenolic) ring or the inner (tyrosyl) ring of the iodothyronine molecule. Outer ring monodeiodination of T4 produces T3, the form of thyroid hormone with the greatest affinity for the thyroid nuclear receptor. Inner ring monodeiodination of T4 produces reverse T3 (rT3), an inactive metabolite. In adults, between 70% and 90% of circulating T3 is derived from peripheral conversion of T4 and 10% to 30% from direct glandular secretion. Nearly all circulating rT3 derives from peripheral conversion, with only 2% to 3% coming directly from the thyroid gland. T3 and rT3 are progressively metabolized to diiodo, monoiodo, and noniodinated forms of thyroxine, none of which possesses biologic activity.
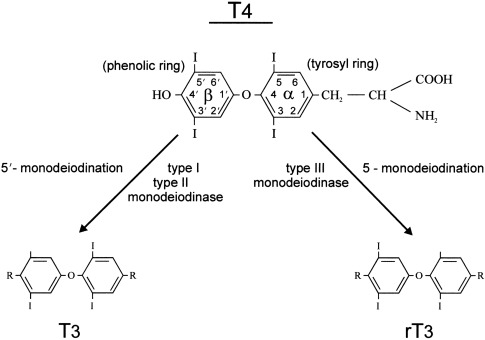
Two types of outer ring iodothyronine monodeiodinases have been described. Type I deiodinase (predominantly expressed in liver, kidney, and thyroid) is a high-Km enzyme inhibited by propylthiouracil and stimulated by thyroid hormone. Type II deiodinase (predominantly located in brain, pituitary, placenta, skeletal muscle, heart, thyroid, and brown adipose tissue) is a low-Km enzyme insensitive to propylthiouracil and inhibited by thyroid hormone. Type I and II deiodinases contribute to circulating T3 production, whereas type II acts to increase local tissue levels of T3 as well. An inner ring deiodinase (type III deiodinase) has been characterized in most fetal tissues, including placenta. This enzyme system catalyzes the conversion of T4 to rT3 and T3 to diiodothyronine. All three deiodinase enzymes are selenoproteins.
Deiodination is developmentally and thyroid-state regulated. In the human fetal brain, type II deiodinase activity in the cortex increases between 13 and 20 weeks’ gestation and by about 50% over the last third of gestation. There is a general inverse correlation of type II and type III activities. Both of these deiodinase species are thyroid hormone responsive.
Fetal thyroid hormone metabolism is characterized by a predominance of type III enzyme activity (particularly in liver, kidney, and placenta), accounting for the increased circulating concentrations of rT3 observed in the fetus. However, the persistence of high circulating rT3 concentrations for several weeks in the newborn indicate that type III deiodinase activity expressed in nonplacental tissues is important. The mixture of type II and type III deiodinase activities in the placenta provides for the conversion of T4 to T3 and of T4 and T3 to rT3 and T2, respectively.
Sulfated iodothyronines are the major thyroid hormone metabolites circulating in the fetus. Sulfokinase enzymes are present early in fetal life, and sulfation of the phenolic hydroxyl group of the iodothyronine molecule may be a normal prerequisite step for monodeiodination. The sulfated iodothyronines are preferred substrates for the type I deiodinase, and concentrations are high in fetal serum in part because of low type I deiodinase activity. However, increased production of sulfated metabolites is also involved. There is evidence that T3S has biologic activity, that is, it suppresses TSH in vivo, suggesting that it can be desulfated by one or more tissue sulfatase enzymes. The low production rates and low levels of T3 metabolites and the high ratio of inactive to active metabolites suggest that fetal thyroid hormone metabolism is largely oriented to inactivating T4, presumably to avoid tissue thermogenesis and to potentiate the anabolic state of the rapidly growing fetus. This is mediated by early activation of type III monodeiodinase, inactivation of type I monodeiodinase, and augmented iodothyronine sulfation.
The developmental expression of type II deiodinase in brain and other tissues provides for local T3 supply to specific tissues (particularly in the event of T4 deficiency) and helps guarantee provision of T3 during gestation, when brain development is thyroid hormone dependent.
Thyroid Hormone Cell Membrane Transporters and Receptors
All thyroid-sensitive cell populations express iodothyronine membrane transporters, which are required for hormone entry into the cell. These belong to families of integrin, organic anion, amino acid, and monocarboxylate solute carriers. The importance of these transporters is highlighted by the role of mutations inactivating human monocarboxylate transporter 8 (MCT8) in an X-linked syndrome of severe psychomotor retardation (previously called the Allan-Herndon-Dudley syndrome ) combined with mild abnormalities of thyroid function, characterized by high T3, low T4, and normal or high TSH. MCT8 is thought to play a role in the entry of T3 into neurons, after deiodination of T4 to T3 in neighboring astrocytes. In addition, MCT8 is involved in the transfer of T3 across the blood-brain barrier. Lastly, the abnormalities in thyroid hormone levels and TSH are also caused by the effect of MCT8 on deiodination.
Thyroid hormone effects are mediated predominantly via nuclear thyroid hormone receptors (TRs), which bind to deoxyribonucleic acid (DNA) to regulate gene transcription. Two mammalian genes code for TR, TRalpha and TRbeta , and alternative mRNA splicing leads to the production of four major thyroid hormone-binding transcripts: TRalpha1, TRalpha2, TRbeta1, and TRbeta2. The TRs exist as monomers, homodimers, and heterodimers with other nuclear receptor family members, such as the retinoid X receptors. TRalpha1 is the predominant subtype in bone, gastrointestinal tract, heart, and brain. TRbeta1 is expressed in liver, kidney, heart, lung, brain, cochlea, and pituitary. TRbeta2 is expressed in pituitary, retina, and cochlea. The receptors function redundantly, as indicated by knockout studies in mice, but predominant effects of one or another TR have been described.
In humans, the specific roles of TRalpha and TRbeta are illustrated by the phenotypes observed in patients with inactivating mutations in the corresponding genes. The syndrome of thyroid hormone resistance initially described in 1967 was later found to be caused by mutations inactivating TRbeta that occur either de novo or are inherited in an autosomal dominant fashion; however, in some patients with thyroid hormone resistance, TRbeta is normal and the molecular defect remains elusive. Mutations in TRalpha have also been described, occurring de novo in one patient and transmitted from father to daughter in one pedigree.
Thyroid hormone-programmed development of fetal tissues requires the interaction of local tissue monodeiodinase I and II, TRs, thyroid receptor coactivators, and thyroid-responsive genes. In most responsive tissues, the timing of maturation events is controlled by the TRs acting as a molecular switch. In the absence of T3, the unliganded receptor recruits corepressors, thereby repressing gene transcription. Local tissue maturation events are stimulated by the coincident availability of T3, liganded T3 receptor, T3-mediated receptor exchange of corepressors with coactivators, and activation of responsive gene transcription.
In the human fetus, low levels of TR binding have been detected in brain tissue at 10 weeks’ gestational age—and liver, heart, and lung TR binding is observed at 16 to 18 weeks. TR levels in human fetal cerebral cortex and cerebellum increase markedly during the second and third trimesters. Information is limited regarding the timing of appearance of thyroid hormone tissue effects in the human fetus.
The birth length of the athyreotic human neonate is within normal limits: the linear growth of the human fetus is programmed independently of thyroid hormones by a complex interplay of genetic, nutritional, and hormonal factors, as well by mechanical uterine constraint. However, 50% to 60% of athyreotic newborns manifest delayed epiphyseal maturation and have large fontanelles. In addition, neonates with severe CH may have large fontanelles, macroglossia, umbilical hernia, prolonged jaundice, and feeding difficulties. However, the classic clinical manifestations of CH appear progressively only during the early months of life. These include myxedema, a slow linear growth, and delayed attainment of psychomotor milestones, which can be mitigated by early diagnosis through neonatal screening programs with institution of thyroid hormone replacement. The normal IQ of athyreotic infants treated early through newborn screening seems attributable to T4 of maternal origin coupled with upregulation of type II monodeiodinase in fetal brain tissue in the face of low fetal serum T4.
Postnatal thermogenesis is mediated via the brown adipose tissue prominent in subscapular and perirenal areas in the mammalian fetus and neonate. Heat production in brown adipose tissue is stimulated by catecholamines via beta-adrenergic receptors and is thyroid hormone dependent. The uncoupling protein thermogenin unique to brown adipose tissue is located on the inner mitochondrial membrane and uncouples phosphorylation by dissipating the proton gradient created by the mitochondrial respiratory chain. The type II monodeiodinase in brown adipose tissue mediates local T4 to T3 conversion. Full thermogenin expression in brown adipose tissue requires both catecholamine and T3 stimulation. Brown adipose tissue matures progressively in the fetus but remains thermoneutral until stimulated by catecholamines in the perinatal period. Brown adipose tissue thermogenesis is immature in small premature infants, and brown adipose tissue mass decreases in the neonatal period in full-term infants, as the capacity for nonshivering thermogenesis develops in other tissues. Uncoupling protein-2 is found in many tissues but does not appear to be regulated by beta-adrenergic agonists or thyroid hormone. Uncoupling protein-3 is expressed in muscle and white adipose tissue, as well as brown adipose tissue. Muscle uncoupling protein-3 is regulated by beta3-adrenergic stimulation and thyroid hormone and presumably contributes to nonshivering thermogenesis in humans. mRNA levels for uncoupling protein-3 are also regulated by dexamethasone, leptin, and starvation, but the regulation differs in brown adipose tissue and muscle.
The critical role of thyroid hormones in central nervous system (CNS) maturation has long been recognized. Nervous system development involves neurogenesis, gliogenesis, neural cell migration, neuronal differentiation, dendritic and axonal growth, synaptogenesis, myelination, and neurotransmitter synthesis. Thyroid hormones have been shown to stimulate several developmentally regulated nervous tissue genes, but the role of these factors in the CNS developmental program remains undefined. Available evidence suggests that deficiency or excess of thyroid hormones alters the timing or synchronization of the CNS developmental program, presumably by initiating critical gene actions or other genetic CNS maturation events.
Perinatal Changes in Thyroid Function
After delivery, the neonate must rapidly convert from the fetal state of predominant thyroid hormone inactivation to a state of relative thyroidal hyperactivity. During the first hours after birth, there is an abrupt increase in circulating T4 and T3 levels. This is caused by the abrupt increase in hypothalamic TRH and pituitary TSH secretion stimulating increased thyroid hormone secretion. As mentioned earlier, the cold-stimulated TRH-TSH surge is short-lived and mean TSH concentrations decrease progressively to normal infant levels by 3 to 5 days.
Serum T3 levels increase in response to the TSH surge, because of stimulation of thyroidal T3 secretion and of a combined cortisol- and T4-stimulated increase in hepatic type I deiodinase activity. Placental separation decreases T3 deiodination (to inactive T2), contributing to the early postnatal increase in serum T3 concentration. The type II deiodinase activity in brown adipose tissue increases during the last weeks of gestation to potentiate catecholamine-stimulated brown adipose tissue thermogenesis, thereby contributing to the maintenance of the body temperature of the neonate.
Congenital Hypothyroidism
Newborn Screening
CH is one of the most common causes of preventable intellectual disability. Its prevalence by clinical ascertainment was around 1/6700. By biochemical screening, the estimated prevalence predictably depends on the screening method and cutoffs and on how the diagnosis is confirmed, but it has been reported to be as high as around 1/1000 in some geographic areas. Newborn screening was first established for phenylketonuria and 10 years later for CH, by measuring either total T4 or TSH. It has now been adopted throughout the industrialized world and has resulted, over the past half century, in the disappearance of “sporadic cretinism,” an unqualified public health success. Typically, a blood sample is collected by heel-prick, blotted on filter paper (the “Guthrie card”) and sent to a central laboratory. Newborns with a positive result (i.e., a low T4 or an elevated TSH) are promptly referred to a specialized center for diagnostic confirmation and for treatment initiation. Fig. 8.6 illustrates the algorithm currently used in Québec, Canada, which has been using TSH as the primary analyte since 1987; in samples with mild/moderately elevated TSH, total T4 is measured to determine the urgency of referral. Initially, total T4 was the primary analyte measured in the United States, but now about half of the states have switched to TSH. The latter approach misses central hypothyroidism, but this condition is at least 10-fold less frequent than primary CH and rarely isolated, so that signs associated with growth hormone, adrenocorticotropin hormone, and luteinizing hormone deficiency (hypoglycemia, cholestatic jaundice, micropenis, and cryptorchidism) lead to the early diagnosis.
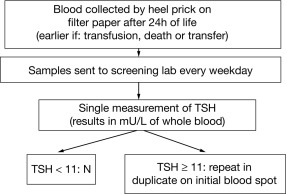
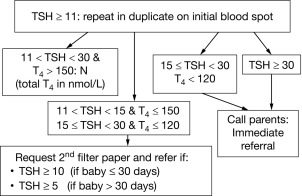
Because of the neonatal TSH surge after birth, the sample for CH screening should be taken after 24 hours of life to avoid an unacceptable number of false positives. Early discharge of babies makes this logistically challenging, and the possibility of using cord blood may have to be revisited. The debate about the optimal TSH cut off to use in screening algorithms is ongoing. The historically used values of 20 to 50 mU/L, to define a positive result, have been lowered in most programs to 6 to 20 mU/L, provided the sample was not taken in the first 24 hours of life. Most additional cases detected have mild and often transient functional disorders (i.e., thyroid anatomy is normal), and whether treatment of these infants impacts developmental outcomes remains to be established. On the other hand, the few cases of CH missed by screening are most commonly caused by human errors in handling samples or in reporting results. Because of fetal blood mixing, truly falsely negative results can occur in the affected twin of a discordant monozygotic pair and obtaining a second screening sample at 14 days in same-sex twins has been advocated. On the other hand, a delayed rise in TSH has been observed, mostly in premature infants, and has led some programs to routinely obtain a second screening sample on all infants. In most such infants, thyroid function ultimately normalizes and there is no evidence of treatment benefit.
From an epidemiological standpoint, the apparent increase in CH prevalence is largely caused by changes in screening cut-offs. In fact, overt permanent CH caused by thyroid dysgenesis confirmed by radionuclide scanning has remained stable despite changes in screening approaches. Predictably, the proportion of cases of CH caused by dyshormonogenesis, a group of autosomal recessive disorders, is higher in populations with high rates of consanguinity. Lastly, a lower overall prevalence of CH in those of African descent compared with Caucasian infants has been observed in the United States and, for CH caused by thyroid dysgenesis specifically, in Montreal, but there are no screening data yet from sub-Saharan Africa. Along this line, it is sobering that only 30% of the world’s children benefit from the benefits of newborn screening. In many low and middle-income countries, transporting Guthrie cards to a central laboratory is unrealistic. Point-of-care testing is likely the way of the future, and measuring devices are being developed.
Thyroid Dysgenesis
The term thyroid dysgenesis encompasses thyroid ectopy, athyreosis, orthotopic hypoplasia, and absence of one lobe, whereas dyshormonogenesis is a group of defects in thyroid hormone synthesis by a normally located and shaped gland ( Table 8.1 ). Dysgenesis underlies 80% to 85% of overt CH and is generally an isolated malformation, except for mild defects in heart septation, which have been found in 2% to 5% of cases.
| Type of Permanent Congenital Disorder | Estimated Birth Prevalence |
|---|---|
Thyroid Dysgenesis:
| 1:5,000 1:15,000 |
| Thyroid Dyshormonogenesis | 1:30,000 |
| Hypothalamic-Pituitary Hypothyroidism | 1:16,000 |
| Thyroid Hormone Resistance | 1:40,000 |
Three-quarters of patients with CH caused by dysgenesis have a sublingual gland. This results from a defect in thyroid migration during embryonic development from the base of the tongue to the anterior neck. On scintigraphy, the only tissue visible is round, lacking the lateral lobes characteristics of orthotopic glands. In about 10%, there is a dumbbell-shaped image, suggesting that some cells have initiated migration and others not. The molecular mechanisms regulating migration of the embryonic thyroid are largely unknown. Ectopic thyroids are fully differentiated and have a normal follicular architecture and the associated CH likely reflects a reduced number of cells (lack of lateral lobes) and limited TSH-induced cell growth. The severity of CH is variable but is stable over time, suggesting normal postnatal survival of the ectopic cells. The 25% of newborns with dysgenetic CH who have no visible uptake of tracer on scintiscan may have true athyreosis, in which cases serum TG is very low. When there is no visible tissue but serum TG is detectable, it seems appropriate to use the term apparent athyreosis , which may result from complete inactivation of the TSH receptor, either from the transplacental passage of blocking immunoglobulins (in which case CH is transient) or from biallelic inactivating mutations of the TSH receptor gene (in which case CH is permanent). Orthotopic thyroid hypoplasia accounts for a smaller percentage of cases of overt CH. Lastly, congenital absence of a thyroid lobe, most often the left, is found in up to one in 500 normal children but may be seen in association with permanent or transient CH.
A genetic susceptibility to thyroid dysgenesis was suggested by the observation that about 1% of affected patients have an affected first-degree relative, giving a 40-fold increase in relative risk overt the population prevalence of one in 4000 for dysgenetic CH. On the other hand, discordance between monozygotic twins is the rule and the three to one female predominance of thyroid ectopy are not compatible with standard Mendelian inheritance. To reconcile these contradictory epidemiological findings, a two-hit model has been proposed, in which thyroid dysgenesis requires the association of a germline susceptibility variant with a postzygotic event occurring at the somatic level, in developing thyroid tissue. Because ectopic thyroids seldom need to be surgically removed, most of the work of the past 20 years has focused on leukocyte DNA, but disease-causing mutations have been found in a small percentage of patients with dysgenetic CH and in only a handful of those with the most common phenotype, thyroid ectopy. Clinically, a DNA diagnosis only needs to be sought if there are suggestive extrathyroidal manifestations or a family history of dominant or recessive transmission of dysgenetic CH ( Table 8.2 ). In addition, generally mild CH, mostly associated with left hemithyroid, appears to occur more frequently in Di George and Williams syndrome than in the general population. In Di George syndrome, the deleted region on chromosome 22q11 generally encompasses TBX1 , a gene involved in vessel development, further emphasizing the link between the development of the thyroid and of the cervical vasculature.
| Gene | Transmission | Thyroid Phenotype | Other Features |
|---|---|---|---|
| TSHR | AR | From apparent athyreosis to normally appearing gland | None |
| NKX2.1 | De novo or AD | From apparent athyreosis to normal gland, usually mild ↑ TSH |
|
| FOXE1 | AR | True athyreosis |
|
| Pax8 | AD or de novo | From apparent athyreosis to normally appearing gland | Cysts within thyroid remnants |
| GLIS3 | AR | From apparent athyreosis to normally appearing gland |
|
| NETRIN | de novo | Ectopy | Ventricular septal defect |
| JAG1 | de novo | Ectopy, apparent athyreosis, orthotopic hypoplasia | Alagille syndrome |
| BOR | AR | Ectopy, athyreosis | None |
| TUBB1 | AR | Ectopy, orthotopic hypoplasia, hemithyroid | Macroplatelets |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


