Chapter Outline
Etiology: Membrane Protein Defects
Conditions That Camouflage Hereditary Spherocytosis
Animal and Fish Models of Hereditary Spherocytosis and Elliptocytosis
HEREDITARY ELLIPTOCYTOSIS AND PYROPOIKILOCYTOSIS
STOMATOCYTOSIS, CRYOHYDROCYTOSIS, AND XEROCYTOSIS
Overhydrated Hereditary Stomatocytosis
Other Hereditary Stomatocytosis Syndromes
Classification Based on the Temperature Dependence of Cation Leaks
ACQUIRED DISORDERS OF RED CELL MEMBRANE LIPIDS
During erythropoiesis the red cell membrane responds to erythropoietin and imports the billion or so iron atoms each red cell needs to complete hemoglobin synthesis. It sequesters the reductants required to protect hemoglobin and other cell proteins from corrosion by oxygen, and selectively retains other vital components such as organic phosphates, manufactured with precious adenosine triphosphate (ATP) energy, but it lets metabolic detritus escape. It even helps regulate metabolism by reversibly binding and inactivating many glycolytic enzymes. In the circulation it carefully balances cation and water concentrations so red cells do not shrivel or burst. Simultaneously, it exchanges tremendous numbers of bicarbonate and chloride anions (≈10 to 30 billion/second/red cell), which aids transfer of carbon dioxide from the tissues to the lungs. It also maintains a slippery exterior so that red cells do not adhere to endothelial cells or aggregate and clog capillaries. Finally, buttressed by the “membrane skeleton,” a protein scaffold that lines the inner membrane surface, the membrane achieves the critical combination of strength and flexibility needed to survive four months in the circulation. Failure of any of these, or numerous other, functions shortens red cell survival, a process termed hemolysis. In fact, all hemolysis is ultimately due to membrane failure. This chapter will focus on inherited disorders of red blood cell membranes, particularly hereditary spherocytosis and hereditary elliptocytosis, the two most common and best understood diseases.
Hereditary Spherocytosis
Hereditary spherocytosis (HS) ( Table 16-1 ) is a common, inherited hemolytic anemia in which defects of spectrin or of proteins that attach spectrin to the membrane—ankyrin, protein 4.2, or band 3—lead to spheroidal, osmotically fragile cells that are selectively trapped in the spleen, resulting in a shortened red cell life span.
| Common Conditions |
| Hereditary spherocytosis |
| Immunohemolytic anemias (warm antibody type) |
| ABO hemolytic disease in neonates † |
| Uncommon to Rare Conditions |
| Hereditary stomatocytosis † |
| Hereditary cryohydrocytosis † |
| Clostridial sepsis |
| Hemolytic transfusion reactions |
| Severe burns and other red cell thermal injuries |
| Spider, bee and snake venoms † |
| Severe hypophosphatemia † |
| Acute red cell oxidant injury (e.g., glucose-6-phosphate dehydro-genase deficiency during hemolytic crisis, Wilson disease) † |
| Congenital dyserythropoietic anemia, type II † |
* Some spherocytes may be seen in other diseases where they are not the predominant red cell morphology (e.g., hereditary pyropoikilocytosis, Heinz body hemolytic anemias, severe liver disease, microangiopathic hemolysis, hexokinase deficiency).
† Spherocytes are the predominant morphology in only a subset of patients.
History
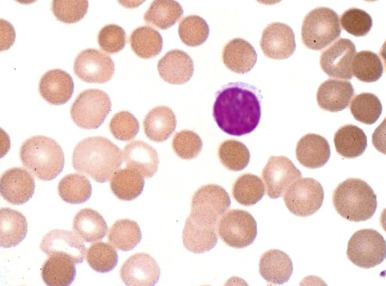
HS was first recognized more than 140 years ago by two Belgian physicians, Vanlair and Masius, who gave a remarkably thorough account of the disease. They described a woman who suffered from the clinical symptoms now known as hallmarks of HS—anemia, jaundice, splenomegaly, and recurrent abdominal pain. The authors noted that most of the woman’s red cells were spherical ( Fig. 16-1 ) and hypothesized that a combination of splenic enlargement and liver atrophy led to their rapid destruction and the patient’s anemia. They also noted that the patient’s sister had suffered from an identical illness. In the 1890s, the British physicians Wilson and Stanley recognized the hereditary nature of the disease and were the first to note the characteristic pathologic appearance of the spleen engorged with red cells. Subsequently, a report by Minkowski in 1900, in the German literature, received wide attention, and many additional papers soon appeared, including Chauffard’s classic description of osmotic fragility and reticulocytosis as hallmark laboratory features of the disease. The first successful splenectomy for HS was unintentionally performed by Wells in England 20 years before splenectomy became widely accepted and 3 years before Wilson’s description of HS appeared. While operating on a jaundiced woman for a supposed uterine fibroid, Wells encountered and bravely removed an enormous spleen. The patient recovered and her jaundice disappeared. Forty years after the splenectomy performed by Wells, Dawson found the abnormal erythrocyte osmotic fragility during an examination of the woman and her son. The major clinical features of HS were defined by the 1920s, although nothing was known about the pathophysiology of the disease. Readers interested in more details about these and other aspects of the history of HS should consult the chapters by Dacie, Crosby, and Wintrobe in the wonderful book Blood, Pure and Eloquent. Recent reviews of HS are also available.

Prevalence and Genetics
HS occurs in all racial and ethnic groups. It is particularly common in Europeans and their descendants in the New World. The classic estimate is that 1 person in 5000 is affected, but this is almost certainly an underestimate. Surveys of red cell osmotic fragility suggest that mild forms of the disease may be four or five times more common than that. In addition, testing of Swiss patients identified as having hyperchromic red cells on a routine complete blood count led to a diagnosis of HS in 1 of every 1700 individuals, most of whom were asymptomatic. So the prevalence in Europeans is probably closer to 1 in 1500 to 1 in 2000. The disease is also common in Japan and most other countries. Clinical experience suggests it is less common in African Americans and Southeast Asians.
HS exhibits both dominant and nondominant inheritance. About 75% of patients have typical autosomal dominant disease. Homozygotes for dominant HS are very rare, with only a handful of reported cases. The remaining 25% of HS patients show nondominant inheritance. In these patients, both parents are clinically normal but some of them exhibit subtle laboratory abnormalities that suggest a carrier state. The only available molecular survey suggests that the majority of nondominant HS patients have new dominant mutations, which tend to occur at CpG dinucleotides and are associated with small insertions or deletions. This study suggests that true recessive HS is quite rare, perhaps less than 5% of the total, but larger-scale studies are needed.
HS is caused by unique mutations in the membrane skeletal proteins ankyrin, β-spectrin, α-spectrin, band 3, and protein 4.2.
Clinical Presentation
Typical Hereditary Spherocytosis
The typical clinical picture of HS combines evidence of hemolysis (anemia, jaundice, reticulocytosis, gallstones, and splenomegaly) with spherocytosis and a positive family history ( Table 16-2 ; Fig. 16-2 ). Mild, moderate, and severe forms of HS have been defined according to differences in various clinical and laboratory parameters under baseline conditions ( Table 16-3 ). Unfortunately, patients don’t fit neatly into these categories: a person my be classified as “mild” by one criterion and “moderate” by another.
| Clinical Features |
| Pallor |
| Splenomegaly |
| Intermittent jaundice |
| From hemolysis |
| From biliary obstruction |
| Anemic crises: hemolytic, aplastic or megaloblastic |
| Inheritance |
| Dominant ≈ 75% |
| Nondominant ≈ 25% |
| Rare manifestations |
| Leg ulcers |
| Chronic dermatitis |
| Extramedullary hematopoietic tumors |
| Angioid streaks |
| Spinocerebellar degeneration |
| Myocardiopathy |
| Excellent response to splenectomy |
| Laboratory Features |
| Anemia |
| Reticulocytosis |
| Spherocytosis |
| Elevated MCHC and elevated percentage of hyperchromic or hyperdense cells |
| Increased RDW (red cell distribution width, a measure of poikilocytosis) |
| Abnormality in one or more “HS tests” |
| Decreased band 3 (decreased fluorescence of red cells labelled with eosin-5′-maleimide) |
| Increased osmotic fragility (especially in the incubated osmotic fragility test) |
| Abnormal acidified glycerol lysis time or Pink test |
| Abnormal cryohemolysis test |
| Characteristic osmotic gradient ektacytometry profile |
| Normal Coombs (direct antiglobulin) test |
| Decreased red cell spectrin, or spectrin and ankyrin, or band 3 and protein 4.2, or absent protein 4.2 and decreased CD47 |
| Normal | Mild Spherocytosis | Moderate Spherocytosis | Moderately Severe Spherocytosis | Severe Spherocytosis * | |
|---|---|---|---|---|---|
| Inheritance | — | Autosomal dominant | Autosomal dominant, de novo mutation | Autosomal dominant, de novo mutation | Autosomal recessive |
| Proportion of hereditary spherocytosis cases | — | ≈20% to 30% | ≈60% to 70% | ≈10% | <5% |
| Hemoglobin (g/dL) † | 11.5-16 ‡ | 10.5-15 | 8-12 | 6-8 | <6 |
| Reticulocytes (%) † | 0.5-1.5 | 1.5-6 | ≥6 | ≥10 | ≥10 |
| Bilirubin (mg/dL) † , ‖ | 0-1 | 0.5-2 | ≥2 | ≥2 | ≥3 |
| Peripheral smear † | Normal | Mild spherocytosis | Spherocytosis | Spherocytosis | Spherocytosis ± poikilocytosis |
| Osmotic fragility (fresh) | Normal | Normal or slightly increased | Increased | Increased | Greatly increased |
| Osmotic fragility (incubated) | Normal | Usually increased | Increased | Increased | Greatly increased |
| MCHC (g/dL) § | 32-36 | 34-37 | 34-38 | 35-39 | |
| RDW (%) § | 11-14 | 12-19 | 16-23 | 20-30 | |
| Hb/MCHC * | 0.38-0.41 | 0.35-0.40 | 0.29-0.33 | 0.18-0.28 | |
| Hb/RDW § | 0.95-1.05 | 0.7-1.0 | 0.48-0.74 | 0.16-0.35 | |
| Serum transferrin receptor (nmol/L) § | 18-25 | 30-65 | 80-125 | 100-150 | |
| Erythropoietin (mIU/mL) § | 7-16 | 9-30 | 25-90 | 30-300 | |
| Membrane protein patterns (SDS-PAGE) ¶ | — | “Normal” # Slight ↓ spectrin Slight ↓ spectrin and ankyrin Slight ↓ band 3 and 4.2 Absent protein 4.2 and ↓ CD47 | ↓ Spectrin ↓ Spectrin and ankyrin ↓ Band 3 and protein 4.2 Absent protein 4.2 and ↓ CD47 | ↓ Spectrin ↓ Spectrin and ankyrin ↓ Band 3 and protein 4.2 | ↓↓ Spectrin ↓↓ Spectrin and ankyrin ↓↓ Band 3 and protein 4.2 ** |
| Transfusions | — | No | Sometimes required in infancy or with aplastic crisis | Occasionally with crises | Regular * |
| Splenectomy | — | Rarely, partial splenectomy †† | Sometimes; consider partial splenectomy | Usually (6-9 years) | Yes (>3 years) |
* Patients with severe disease are transfusion dependent by definition. Values are in untransfused patients or at nadir before transfusion.
† Data modified from Eber SW, Armbrust R, Schröter W: Variable clinical severity of hereditary spherocytosis: relation to erythrocytic spectrin concentration, osmotic fragility and autohemolysis. J Pediatr 177:409–416, 1990.
§ Ranges shown encompass the majority of individuals in each category. From Rocha S, Costa E, Rocha-Pereira P, et al: Complementary markers for the clinical severity classification of hereditary spherocytosis in unsplenectomized patients. Blood Cells Mol Dis 46:166–170, 2011.
‖ Multiply by 17.1 to convert to µmol/L.
¶ Indicates common patterns observed on SDS gels. Decreased spectrin alone is seen in α-spectrin or β-spectrin defects. Decreased spectrin and ankyrin are observed with ankyrin defects. Decreased band 3 and protein 4.2 occur with band 3 defects. Absent protein 4.2 and decreased CD47 occur with protein 4.2 defects.
# Patients with mild spherocytosis who appear normal probably have small deficits (10% to 15%) that cannot be distinguished from normal findings on SDS gels.
** Rare patients with severe spherocytosis who are homozygous or compound heterozygous for band 3 defects.
†† Consider in adolescents and adults who require a cholecystectomy or have disfiguring chronic jaundice.
Initial assessment of a potential HS patient should include questions about neonatal or subsequent jaundice, anemia, splenomegaly, or gallbladder symptoms; transfusions; and a family history of these things and of splenectomies or cholecystectomies. The physical examination should seek signs of pallor, scleral icterus, and splenomegaly. Pediatricians should also examine the parents and siblings if they are present, particularly for splenomegaly, which is more evident in older patients. Scleral icterus is usually subtle and best perceived in daylight and at the edge of the conjunctivae ( Fig. 16-3 ). Similarly, a spleen that cannot be palpated in a supine patient can sometimes be felt when the patient is placed on his or her right side with knees bent.
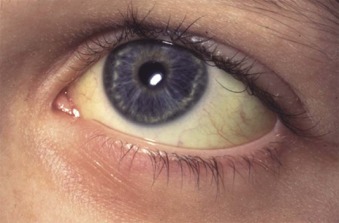
HS typically presents in infancy or childhood, but it may present at any age. In children, anemia is the most common presenting complaint (50%), followed by splenomegaly, jaundice, or a positive family history. No comparable data exist for adults. The majority of patients with HS have incompletely compensated hemolysis and mild to moderate anemia. The anemia is often asymptomatic except for fatigue and mild pallor or, in children, nonspecific parental complaints such as irritability. Jaundice is seen at some time in about one half of patients, usually in association with viral infections. When jaundice is present it is acholuric (i.e., unconjugated [indirect] hyperbilirubinemia without detectable direct bilirubinuria). Palpable splenomegaly is detectable in about one half of infants and most (70% to 95%) older children and adults. Typically, the spleen is modestly enlarged (2 to 6 cm below the costal margin), but it may be massive. There is no proven correlation between the size of the spleen and the severity of HS, although, given the pathophysiology and the response of the disease to splenectomy, such a correlation probably exists.
Typical HS is associated with both dominant and nondominant inheritance. The nondominant disease tends to be more severe, but there is considerable overlap. Clinical severity and the response to splenectomy roughly parallel the degree of spectrin (or ankyrin) deficiency in patients with defects in either protein. Disease in such patients can vary from mild to severe. Patients who are heterozygotes for band 3 defects have typical, moderate HS and moderate band 3 deficiency (15% to 35%). Severe HS is rare in this population unless the patient carries two defective band 3 genes. Protein 4.2 deficiency is also associated with milder disease. Overall, patients with deficiency of spectrin or both spectrin and ankyrin have more severe disease than patients with band 3 or protein 4.2 deficiency, but the differences are not great because true “severe” HS is rare.
In general, members of the same family will have a similar clinical picture, but there are exceptions, presumably due to varying compensation by the normal allele in patients who are heterozygous for a mutant gene, or by the coinheritance of another deleterious allele. Such alleles should be sought in patients whose disease is much more severe than that of the rest of the family.
Silent Carrier State
The parents of patients with “nondominant” HS are clinically asymptomatic and do not have anemia, splenomegaly, hyperbilirubinemia, or spherocytosis on peripheral blood smears. However, some have subtle laboratory signs of HS, including slight reticulocytosis (average, 2.1 ± 0.8%), diminished haptoglobin levels, slightly elevated osmotic fragility, or slightly shortened times in the acidified glycerol lysis test (AGLT). Screening of normal Norwegian and German blood donors with the osmotic fragility test or AGLT showed a 0.9% to 1.1% incidence of previously unsuspected “very mild” HS. Presumably, some of these individuals were silent carriers of recessive HS genes, whereas others just had mild dominant HS. Unfortunately, no systematic molecular survey of this group of patients has been conducted.
Mild Hereditary Spherocytosis
Red cell production and destruction are balanced or nearly balanced in 20% to 30% of patients with HS. These persons are considered to have “compensated hemolysis” and are usually asymptomatic. In some patients, the diagnosis may be difficult because hemolysis, splenomegaly, and spherocytosis are unusually mild. In this group of patients, reticulocyte counts are generally less than 6%, and only 60% of patients have obvious spherocytosis on peripheral blood smears. About 10% of HS patients have very few (≤2%) or no detectable spherocytes. In addition, red cell spectrin and ankyrin levels are typically more than 80% of normal. As noted, many of these patients have band 3 or protein 4.2 defects.
Hemolysis may become severe with illnesses that cause splenomegaly, such as infectious mononucleosis. Hemolysis may also be exacerbated by pregnancy or exercise, to the point where it may impair athletic performance in endurance sports. In many of these patients, HS is diagnosed during family studies or discovered when adult patients are diagnosed with splenomegaly or gallstones. Although mild HS is usually familial, it develops sporadically in families with more severe disease. Presumably this is due to the coinheritance of modifying genes, such as those affecting spectrin or ankyrin synthesis or splenic function.
How to Explain “Compensated Hemolysis”
It is unclear why patients with HS and “compensated hemolysis” (i.e., normal hemoglobin levels) continue to have erythroid hyperplasia. Erythroid stimulation should disappear once hemoglobin levels reach normal if hemoglobin functions normally, as it does in HS. That it does not is shown by the fact that erythropoietin is inappropriately elevated (up to eight times normal) in HS patients with compensated hemolysis and no anemia, and the fact that the serum transferrin receptor concentration, a measure of erythropoiesis, is elevated. The levels of 2,3-diphosphoglycerate (2,3-DPG) are reportedly low in hereditary spherocytes before splenectomy, which could explain it; however the P 50 of blood from HS patients is normal, which does not make sense. A more likely possibility is that the dehydrated HS red cells are rheologically impaired and do not adequately perfuse the juxtaglomerular renal vessels, where erythropoietin is produced, even when the hematocrit is normal. If this hypothesis is correct, then erythropoietin production should correlate inversely with red cell deformability in any disorder where red cells are dehydrated (e.g., sickle cell anemia, hereditary xerocytosis). Clinically, the high level of erythropoietin and persistent erythroid hyperplasia in patients with HS and a normal hemoglobin level indicate that physiologically the patients are still anemic. Indeed, it is possible that all HS patients are physiologically more anemic than their hemoglobin values suggest.
Moderate and Severe Hereditary Spherocytosis
A small fraction of patients with HS (5% to 10%) have moderately severe to severe anemia. Patients with “moderately severe” disease typically have a hemoglobin value of 6 to 8 g/dL, a reticulocyte count of about 10%, a bilirubin level greater than 2 mg/dL, and 40% to 70% of the normal red cell spectrin content. The mean corpuscular hemoglobin concentration (MCHC) is high and the red cell distribution width (RDW) is very high (typically >20%) (see Table 16-3 ). Such patients are more susceptible to the dangers of hemolytic and aplastic crises (see later) and may occasionally need a transfusion. This category includes patients with both dominant and recessive HS and a variety of molecular defects in spectrin and ankyrin. Occasional patients with band 3 defects may fall into this category.
Patients with “severe” disease, by definition, have life-threatening anemia and are transfusion dependent. They often have recessive HS. Most of these patients probably have isolated, severe spectrin deficiency (<40% of normal) resulting from a defect in α-spectrin, but some have ankyrin defects or are homozygous or compound heterozygous for band 3 mutations. Patients with severe HS are also distinguished by red cell morphologic findings. They often have some irregularly contoured or budding spherocytes and bizarre poikilocytes, in addition to typical spherocytes. Such cells are rare before splenectomy in patients with less severe disease, although some may be seen after splenectomy. In addition to the risks of recurrent transfusions, patients with “severe” disease often suffer from an aplastic crisis and may develop growth retardation, delayed sexual maturation, or aspects of thalassemic facies.
Hereditary Spherocytosis in Pregnancy
In general, unsplenectomized patients with HS have no significant complications during pregnancy except for anemia, which worsens because of plasma volume expansion and sometimes because of increased hemolysis. Hemolytic crises during pregnancy requiring transfusion have been reported. Folic acid deficiency is also a risk. One group reported that 20% of patients with HS received transfusions during pregnancy, but in our experience, pregnant patients with HS rarely need transfusions.
Hereditary Spherocytosis in the Neonate
HS often presents as jaundice in the first few days of life. Roughly one half of all patients with HS have a history of neonatal jaundice, and 91% of infants discovered to have HS in the first week of life are jaundiced (bilirubin level >10 mg/dL). In a French study of 402 jaundiced neonates requiring phototherapy, 1% were found to have HS. Hyperbilirubinemia usually appears in the first 2 days of life and bilirubin levels may rise rapidly, driven by the combination of hemolysis and the reduced capacity of the neonatal liver to conjugate bilirubin. Newborns with both HS and Gilbert syndrome, a common polymorphism in the promoter region of the uridine diphosphate–glucuronosyltransferase gene (UGT1A1) (see Chapter 4 ), develop neonatal jaundice more frequently and may develop severe hyperbilirubinemia. Kernicterus is a risk and exchange transfusion or treatment with barbiturates are sometimes necessary, but in most patients the jaundice is controlled with phototherapy.
Only 43% of neonates with HS are anemic at birth (hemoglobin level <15 g/dL), and severe anemia is rare, but those who present at birth tend to have a more severe course. Most have a normal hemoglobin level at birth, which then decreases sharply over the first 3 weeks of life, leading to a transient, severe anemia. In up to three fourths of HS infants, the anemia is severe enough to warrant transfusion or treatment with erythropoietin (see below). This anemia appears to be aggravated by an inability of the infant’s bone marrow to mount an appropriate erythropoietic response to anemia and to the development of splenic function.
Hydrops fetalis has been reported in a few patients and is associated with homozygosity or compound heterozygosity for spectrin or band 3 defects. If hydrops fetalis is detected in utero, the fetus may require intrauterine transfusions. No instances of hydrops fetalis have been associated with ankyrin deficiency. Perhaps infants with ankyrin defects, similar to ankyrin-deficient nb/nb mice, are partially protected in utero by the expression of ankyrin-related proteins in embryonic and fetal erythroblasts.
The diagnosis of HS is sometimes more difficult in the neonatal period than later in life. Splenomegaly is uncommon—at most the spleen tip is palpable—and reticulocytosis is variable and usually not severe. Only 35% of affected neonates have a reticulocyte count greater than 10%. In addition, the haptoglobin level is not a reliable indicator of hemolysis during the first few months of life. An even greater problem is that 33% of neonates with HS do not have significant numbers of spherocytes on their peripheral blood smears. Moreover, because fetal red cells are more osmotically resistant than adult cells when fresh and more osmotically sensitive after incubation at 37°C for 24 hours, the osmotic fragility test occasionally gives false-positive (incubated) or false-negative (unincubated) results unless fetal controls are used. Fortunately, these results have been published, and they appear to reliably differentiate neonates with HS, particularly when the incubated osmotic fragility test is used. However, in our experience it is rarely necessary to use fetal controls. The standard osmotic fragility test usually suffices to make the diagnosis in conjunction with the blood smear, Coombs test, and other data. Tests such as the eosin-5′-maleimide–binding test, AGLT, Pink test, and cryohemolysis test and osmotic gradient ektacytometry (all discussed later) are more sensitive and specific than the osmotic fragility test in older children and adults, but they have not been systematically tested in the neonatal period. Studying the erythrocytes from the parents is also very useful in the diagnosis of HS, particularly if the infant has received a transfusion.
However, it is clear that many infants with HS are not detected in the neonatal period on clinical grounds alone, even infants with hyperbilirubinemia, probably because the disease is not considered or is not obvious. Ascertainment is greatly enhanced if all infants with a high MCHC and a high RDW are evaluated for HS.
As noted above, some infants with HS become progressively more anemic in the first month or two of life and require transfusion. Fortunately, the problem is transient, except in rare patients with severe HS, and usually remits after one or two transfusions. Almost all infants with HS will outgrow the need for transfusion by the end of the first year of life. If the child is otherwise well, we allow the hemoglobin level to fall to 5.5 to 6.5 g/dL before giving transfusions to try to stimulate the marrow and only raise the hemoglobin level to 9 to 11 g/dL after transfusion to avoid suppressing the desired marrow response.
Administration of recombinant human erythropoietin (rHuEpo) to infants with HS was beneficial in reducing blood transfusions in an uncontrolled, open-label study. In this study, 13 of 16 infants given weekly subcutaneous injections of rHuEpo at a dosage of 1000 IU/kg had increases in the absolute reticulocyte count and hemoglobin values. The infants were given the same dosage of rHuEpo weekly based on their initial weight (at a range of initial ages from 16 to 119 days), a weekly hemoglobin level of less than 8 g/dL, and an absolute reticulocyte count of 200 × 10 9 /L or less. Subsequent studies show similar effectiveness with different dosages and different frequencies. Therefore erythropoietin therapy in infants with HS has become an alternative to transfusion, although weekly erythropoietin injections can be challenging for parents. Also, erythropoietin is twice as expensive as a single transfusion and erythropoietin-treated infants don’t always avoid a transfusion. Further studies of recombinant erythropoietin therapy for neonatal anemia in HS are needed to determine the optimal time to initiate treatment, the optimal dosage, and the optimal duration of therapy.
Close observation of infants with HS is necessary to avoid complications of severe anemia. All infants with HS should have hemoglobin and reticulocyte measurements at least monthly during the first 6 months of life to avoid unnecessary side effects of severe anemia. The intervals may be increased thereafter, depending on the severity of anemia, but care must be taken to instruct parents on the signs and symptoms of an aplastic crisis.
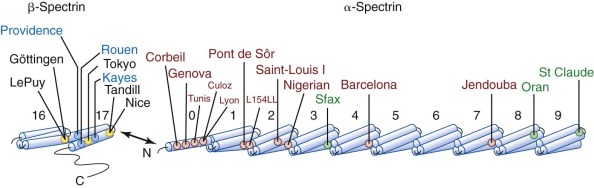
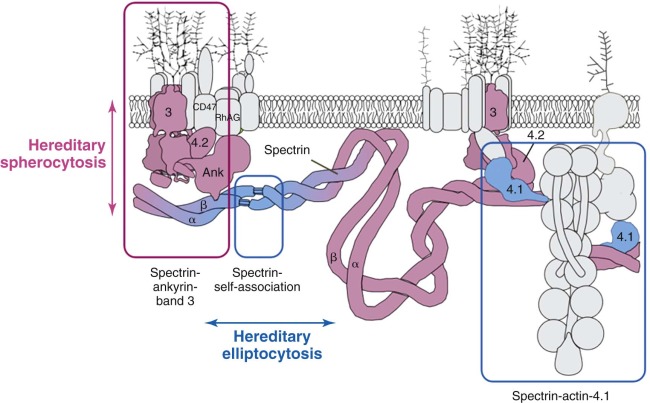
Etiology: Membrane Protein Defects
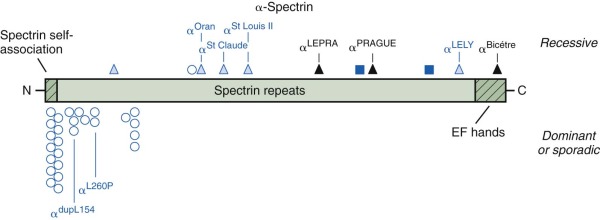
The primary molecular defects in HS reside in membrane skeleton proteins, particularly the proteins whose vertical interactions connect the membrane skeleton to the lipid bilayer: spectrin, ankyrin, protein 4.2, and band 3 ( Fig. 16-4 ). Because each of these proteins has more than one vertical attachment (ankyrin to RhAG as well as band 3, protein 4.2 to CD47 as well as band 3, spectrin to phospholipids and to the band 3 complex near actin), missense mutations that only impair a single protein-protein interaction are not pathologic, whereas reductions in the amounts of the four proteins are. In contrast, missense mutations predominate in hereditary elliptocytosis, which is caused by diminished horizontal interactions between individual proteins that hold the membrane skeleton together, especially defects in spectrin self-association (see Fig. 16-4 ).
Patterns of Membrane Protein Deficiency in Hereditary Spherocytosis
Four patterns predominate: isolated, partial spectrin deficiency, seen with defects in α-spectrin or β-spectrin; combined, partial spectrin and ankyrin deficiency, seen with primary ankyrin defects; combined, partial band 3 and protein 4.2 deficiency, seen with defects in band 3; and nearly complete loss of protein 4.2 and CD47, seen in primary defects of protein 4.2. In Europeans and Americans ankyrin and band 3 defects are most common, roughly 45% and 30%, respectively; β-spectrin defects account for perhaps 20%. Protein 4.2 and α-spectrin defects are both recessive and are rare. However, the pattern of membrane protein defects varies in different populations. In Japan, deficiencies of band 3 and protein 4.2 are the most common forms of HS. In Korean patients, ankyrin and spectrin defects are most common, but protein 4.2 deficiency occurs more often than in European or American patients.
Because most mutations are unique within each family and the mutation itself does not usually predict the phenotype, it is rarely clinically useful to determine the specific molecular defect in patients with HS. Readers who are interested in tables listing many of the mutations described to cause HS should consult lists in various chapters and reviews, including the previous edition of this text, or one of the two versions of the Human Gene Mutation Database: HGMD Professional, a subscription database that is available through many medical libraries, and the public version of HGMD, which is free but lags 3 years behind the subscription version.
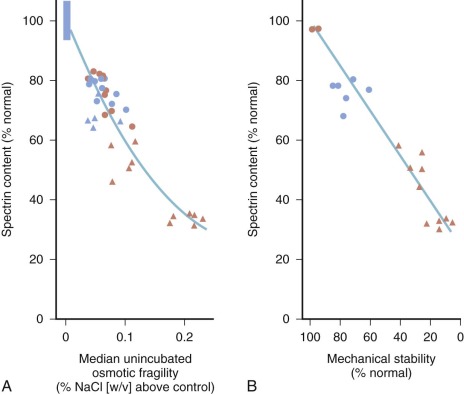
For patients whose red cells are spectrin deficient, the degree of deficiency correlates well with the spheroidicity of HS red cells, the severity of hemolysis, and the response to splenectomy ( Fig. 16-5, A ). The mechanical properties of the cells, particularly their ability to withstand shear stress, also correlate with their spectrin content (see Fig. 16-5, B ). Microscopically, HS red cells with spectrin deficiency show fewer spectrin filaments interconnecting spectrin–actin–protein 4.1R junctional complexes, but overall skeletal architecture is preserved, except in the most severe forms of the disease.
α-Spectrin Defects: Recessive Hereditary Spherocytosis
In humans, α-spectrin synthesis exceeds β-spectrin synthesis by about four to one. Heterozygotes for α-spectrin synthetic defects still produce enough normal α-spectrin chains to pair with all of the β chains that are made. Thus α-spectrin defects are only apparent in the homozygous or compound heterozygous state and exhibit recessive inheritance. Moreover, there may only be a limited spectrum of mutations that cause clinical disease. Production-defective or “thalassemia-type” mutants that only moderately affect expression, such as α-spectrin LELY (to be discussed under hereditary elliptocytosis), have no phenotype, even in the homozygous state. Based on the few reported cases, it appears that the output of the α-spectrin gene must be less than 25% of normal to cause disease and may be as low as 8% of normal and still be compatible with life. No homozygous null mutations have been reported, so it is possible they are lethal in the embryo.
To date, only 2% of all the mutations reported to cause HS involve α-spectrin ( Fig. 16-6 ). Wichterle et al reported a patient with severe HS who was a compound heterozygote for two different α-spectrin gene defects: in one allele, there was a splicing defect associated with an upstream intronic mutation, α LEPRA (low-expression allele Prague); in the other allele, there was a null mutation, α PRAGUE . The α LEPRA allele produces one-sixth as much correctly spliced α-spectrin mRNA as normal. It is a relatively common allele, estimated to be 5% among Caucasians, and is in linkage dysequilibrium with α Bughill , which is an innocent amino acid polymorphism (Ala970Asp). Delaunay reported a similar family combining α LEPRA /α Bughill on one chromosome with a double mutant α LELY /α Bicêtre on the other, where the latter mutation essentially abolished α-spectrin output. In both cases the combination of the severely impaired α LEPRA allele with a null mutation of α-spectrin in trans , leads to significant, spectrin-deficient spherocytic anemia. As expected, even in the most severe forms of α-spectrin–linked recessive HS, obligate heterozygotes are asymptomatic and have little or no spectrin deficiency.
Clinical Features.
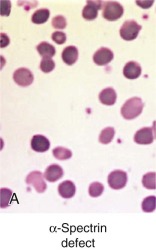
Less than 5% of patients with HS have primary defects in α-spectrin, but most of those patients have severe HS. Their red cells only contain 25% to 50% of the normal amount of both spectrin chains ( Table 16-4 ). Many are transfusion dependent, and even after splenectomy they may experience only a partial recovery. Blood smears are more bizarre than is typical for HS and show intense spherocytosis and irregular contracted cells ( Fig. 16-7, A ).
| <5% of hereditary spherocytosis |
| Autosomal recessive since four-fold more α-spectrin is made than β-spectrin |
| A low expression, “thalassemia-type” defect in α-spectrin is often paired with a second presumably null allele on the opposite chromosome (in trans ). The combination must reduce α-spectrin expression by at least 75% to impair αβ-spectrin assembly enough to cause HS. Homozygous null mutations may be lethal |
| Severe hemolysis and anemia, often transfusion dependent |
| 50%-75% spectrin deficiency in red cells |
| Red cell morphology not well defined but dense, irregular poikilocytes as well as spherocytes in some patients |


β-Spectrin Defects: Dominant Hereditary Spherocytosis
β-Spectrin synthesis limits formation of αβ-spectrin heterodimers, so even modest defects in the expression of β-spectrin are apparent in the heterozygous state and exhibit a dominant pattern of inheritance. Of the reported HS mutations 17% are in β-spectrin, a finding that fits with the general impression that β-spectrin defects account for 15% to 25% of cases. Some groups report a much higher proportion of HS patients with isolated spectrin deficiency when sodium dodecyl sulfate (SDS) gels are used for membrane protein analysis, but that may reflect problems with assessing ankyrin deficiency on SDS gels (see later). The characteristics of HS due to β-spectrin deficiency are listed in Table 16-5 .
| 15%-25% of hereditary spherocytosis cases |
| Almost all individual, dominant null mutations |
| Spectrin content decreased 15%-40% |
| Mild to moderately severe anemia |
| Blood smear with spherocytosis plus 5%-15% spiculated red cells in most patients |
| Spherocytosis plus elliptocytosis (i.e., spherocytic elliptocytosis) in some patients with C-terminal mutations that impair spectrin self-association and decrease spectrin content |
The described mutations in β-spectrin include initiation codon disruptions, frameshift and nonsense mutations, gene deletions and rearrangements, and splicing defects ( Fig. 16-8 ) . Most patients have defects that impair or abolish β-spectrin production from the affected allele. All of the β-spectrin is generated from the normal allele in trans , which compensates to varying degrees, so that the red cell spectrin content is only decreased by 15% to 40% instead of the expected 50%. This suggests that β-spectrin is also synthesized in excess, though much less so than α-spectrin. The degree of compensation probably accounts for much of the variation in clinical severity among patients with β-spectrin HS.
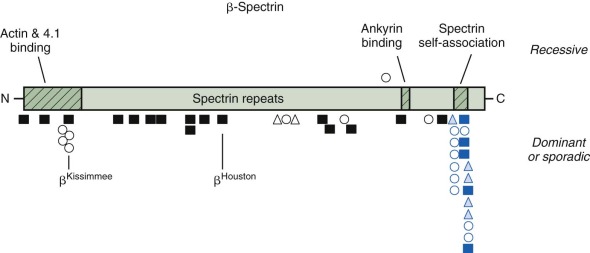
Patients with deletions of the whole β-spectrin locus, or with gross rearrangements, also have autism and learning disabilities, indicating that genes close to β-spectrin on chromosome 14 are involved in cognition. There are HS patients with potentially instructive missense mutations, including a group clustered in the actin and protein 4.1R binding domain (spectrins Atlanta, Kissimmee, and Oakland ) and in the spectrin repeat domain (spectrins Columbus, Birmingham, and Sao Paulo ). It would be interesting to know whether these amino acid substitutions cause HS because they impair a specific function or whether the missense mutations simply destabilize β-spectrin and act like null mutations. In the case of spectrin Kissimmee (Trp202Arg), which is the only mutation where function was examined, both things were true. The mutant protein lacked the ability to bind protein 4.1R and, as a consequence, bound poorly to actin, but it was also unstable and decreased in amount.
All of the reported defects of β-spectrin are limited to single families with the exception of spectrin Houston , a null mutation that has been found in several unrelated kindreds.
Clinical Features.
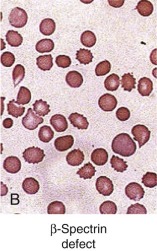
Patients with HS caused by β-spectrin mutations have hemolysis and anemia that vary from mild to moderately severe (see Table 16-5 ). Interestingly, in HS patients in whom peripheral blood morphologic characteristics have been described, those with β-spectrin mutations have had a small population (5% to 15%) of acanthocytes or spiculated spherocytes (spheroechinocytes) as well as spherocytes (see Fig. 16-7, B ).
Ankyrin Defects
Spectrin heterodimers are only stable when bound to the membrane, and ankyrin, the high-affinity binding site, is normally present in limiting amounts. As a result, ankyrin defects are typically expressed as a dominant defect, although recessive mutations have been described. Ankyrin mutations are estimated to account for 30% to 60% of HS cases in Northern European populations and 5% to 10% of HS cases in Japan. Of all the known HS mutations, 35% are Ank1 defects. The characteristics of HS due to ankyrin defects are listed in Table 16-6 .
| 30%-60% of hereditary spherocytosis in the United States and Europe; 5%-10% in Japan |
| Autosomal dominant and autosomal recessive |
| Dominant mutations are mostly null mutations and are nearly all unique |
| Recessive mutations are missense and promoter defects: − 108T→C promoter mutation is common |
| Both spectrin and ankyrin are equally decreased by 15%-50% |
| Mild to moderately severe anemia. Occasionally causes severe disease |
| Blood smear shows spherocytosis without other abnormal morphology |
A role of ankyrin in HS was initially suggested by two patients with an atypical, unusually severe form of HS characterized by bizarre-shaped microspherocytes and red cell spectrin and ankyrin levels that were only 50% of normal. Ankyrin synthesis was half normal; spectrin synthesis was normal, but only 50% of the synthesized spectrin attached to the membrane, presumably owing to the lack of ankyrin sites. These results were the initial indication that ankyrin deficiency or dysfunction leads to concomitant spectrin deficiency in HS.
Additional studies showed that ankyrin deficiency is common in typical, dominant HS. When measured by radioimmunoassay, red cell membrane spectrin and ankyrin levels were less than the normal range in a large fraction of American kindreds, and the two proteins were proportionally decreased in nearly all the deficient kindreds. Similar data have been obtained in multiple populations.
Genetic analyses supported the hypothesis that a defect of the ankyrin gene could cause HS. Initially, studies of atypical patients with HS and karyotypic abnormalities, including translocations and interstitial deletions, defined a locus for HS at chromosomal segments 8p11.2-21.1. After the ankyrin gene was cloned, it was localized to this same region, 8p11.2. Fluorescence-based in situ hybridization of metaphase spreads from a patient with HS and an interstitial deletion of chromosome 8p11.1-p21 provided direct evidence that one copy of the erythrocyte ankyrin gene was deleted. Ankyrin content in the patient’s red blood cell membranes was reduced by 50%. Gene mapping then linked ankyrin to HS in a large family with typical dominant HS. Further studies showed that ankyrin mutations are common in HS and that many HS patients express only one of their two ankyrin alleles.
Ankyrin mutations occur in both dominant and nondominant HS. Frameshift and nonsense mutations, which abolish the normal ankyrin product, are particularly common in dominant HS ( Fig. 16-9 ). They produce mild to moderately severe hemolytic anemia, depending on the output of the remaining normal ankyrin gene and, perhaps, other modifier genes. For example, ankyrin Marburg and ankyrin Einbeck are frameshift mutations that occur in the NH 2 -terminal (membrane) domain. Neither produces a detectable product in mature red cells. They would be expected to have a similar phenotype, but patients with ankyrin Einbeck have very mild disease and normal ankyrin levels, whereas patients with ankyrin Marburg have moderate to severe HS and moderate ankyrin deficiency (64% of normal). Understanding such phenotypic variations is one of the critical issues in HS research.
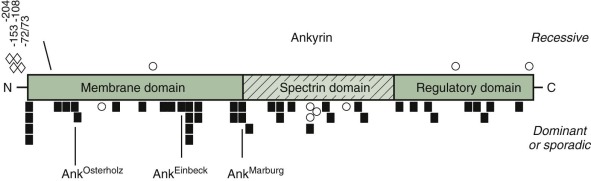
The most common ankyrin variant in patients with recessive HS is a T→C nucleotide substitution in the ankyrin gene promoter −108 bp from the translation start site. The allele occurs in 29% of German HS patients and 3% of normal individuals. It is usually in cis with a G→A nucleotide substitution 153 bp from the start site and impairs ankyrin synthesis in transgenic animals. A −72/73 dinucleotide deletion in the ankyrin promoter also impairs expression by disrupting the recognition site for the TATA-binding protein and interfering with transcription initiation. These “thalassemia-like” promoter mutations are silent in obligate heterozygotes; thus patients with nondominant HS must have a second mutation. The promoter mutations aggravate ankyrin mutations if they are inherited in trans , on the “normal” allele, particularly the −108 mutation, which is relatively common. For example, in a family with ankyrin Osterholtz , which is a null mutation, the daughter, who has a combination of ankyrin Osterholtz /ankyrin −72/73 , has more severe disease than her mother, who only carries ankyrin Osterholtz .
Patients with gene deletions involving the ankyrin locus at 8p11.2 often have features both of HS and Kallmann syndrome (hypogonadotrophic hypogonadism, type 2, due to FGFR1 defects) because the two loci are less than 4 Mb apart. Most of these patients also have psychomotor developmental delay, resulting from loss of another still undefined gene in the region.
Clinical Features.
Peripheral blood smears demonstrate only prominent spherocytosis (see Fig. 16-7, C ). Hemolysis can be mild to severe, and the patients with recessive mutations typically have a more severe clinical course. Almost all dominant ankyrin mutations are private; that is, each individual kindred has a unique mutation. There is one exception, ankyrin Florianopolis , a frameshift mutation that has been identified in HS patients from three unrelated kindreds. Differences in a linked ankyrin gene polymorphism show that the three ankyrin Florianopolis alleles are different and arose independently.
Band 3 Defects: Dominant Hereditary Spherocytosis
Band 3 deficiency is present in approximately 25% to 45% of European and American patients with dominant HS, and band 3 defects account for 37% of the known HS mutations. Erythrocyte membranes from these patients are equally deficient in band 3 and protein 4.2, which requires band 3 to bind to the red cell membrane. The characteristics of HS due to band 3 deficiency are listed in Table 16-7 .
| 25%-45% of hereditary spherocytosis |
| Individual, dominant functionally null mutations. No common defects |
| 15%-40% decrease in band 3 and a similar decrease in protein 4.2 |
| Mild to moderate hemolysis and anemia |
| Spherocytosis with a small number of characteristic button mushroom-shaped or “pincered” cells in most patients |
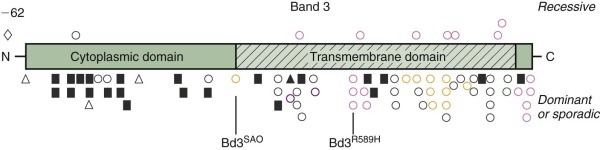
Defects in band 3 produce a variety of red cell phenotypes ( Fig. 16-10 ), including stomatocytosis, cryohydrocytosis, Southeast Asian ovalocytosis (SAO), and acanthocytosis, as well as HS. These are discussed in later sections of the chapter. The HS mutations are spread throughout the band 3 gene, occurring in both the cytoplasmic and membrane-spanning domains. They include nonsense mutations, frameshift mutations, partial gene duplications, other missense mutations, and splicing defects. Many of the mutations cause messenger RNA (mRNA) instability or a misfolded protein that is degraded or not inserted into the membrane. Conserved arginine residues are frequent sites of missense mutations in the transmembrane domain of the band 3 gene. The affected arginine residues are all located in the same position on the inside edge of membrane-spanning segments and are believed to interact with negatively charged phospholipids in the inner bilayer and help orient the segments. As predicted, in vitro studies show that these arginine substitutions, as well as several other HS-associated band 3 missense mutations, exhibit defective cellular trafficking from the endoplasmic reticulum to the plasma membrane, perhaps due to misfolding. Determination of the structure of the cytoplasmic domain of band 3 and detailed studies of the topography of the transmembrane domain are beginning to provide insights into the biologic significance of the missense mutations.
Similar to the other molecular defects that cause HS, band 3 mutations are unique familial defects with no common mutations among affected groups.
Clinical Features.
Clinically, most patients with band 3 mutations have mild to moderate hemolysis and anemia. The band 3 protein is reduced by 15% to 35% in HS red cells. On average, band 3 defects are somewhat milder than ankyrin or β-spectrin defects, although there is considerable overlap in severity. Patients with HS and band 3 deficiencies have a small number (≤1%) of button mushroom–shaped or “pincered” erythrocytes in addition to spherocytes on peripheral blood smears (see Fig. 16-7, D ). This morphologic characteristic is not observed in the other membrane protein defects.
Distal Renal Tubular Acidosis.
Distal renal tubular acidosis (dRTA) is a disorder characterized by the inability of the kidney to acidify the urine below pH 5.5 in the presence of systemic metabolic acidosis. It is caused by failure of H + secretion in the distal nephron. Patients present with various combinations of failure to thrive, hyperchloremic metabolic acidosis, hypokalemia, weakness, metabolic bone disease, and kidney stones. Because a shortened form of band 3 lacking the first 65 amino acids is expressed in the intercalated cells of the kidney cortical collecting ducts where its anion exchange capability is coupled to H + excretion, defects in band 3 would be expected to produce dRTA, and in fact a number of recessive and dominant missense mutations within the membrane domain of band 3 have been identified in dRTA patients (see Fig. 16-10 ). The dominant defects tend to cluster in the middle of the membrane domain (Arg589His has been described in multiple kindreds) or at the C-terminus (see Fig. 16-10 ). The recessive mutations are localized throughout the membrane domain. The dominant mutations seem to be more prevalent in Europe, whereas the recessive mutations are more prevalent in Southeast Asia, but this may be because SAO (band 3 SAO or band 3 Δ400-408 ) is very common in that part of the world (discussed later) and causes dRTA when it is coupled with a recessive dRTA allele.
Surprisingly, few patients with HS due to band 3 defects have dRTA. There are a few exceptions in patients who are homozygous or compound heterozygous for band 3 defects ; however, the majority of HS patients with mutations in band 3 do not have dRTA. Conversely, few patients with band 3 defects causing dRTA have hemolysis or spherocytosis. In general, the dRTA mutations do not impair anion exchange. They cause band 3 to either be retained in the endoplasmic reticulum or Golgi apparatus, or to be mistrafficked to the wrong membrane surface in polarized cells. Most of the transport and trafficking studies have been done in oocytes, Madin-Darby canine kidney (MDCK) cells, or other epithelial cells and not in erythroid cell lines, so it is not possible to test whether band 3 trafficking is aberrant in kidney cells but not in red cells. This would explain why the dRTA mutants do not cause HS, and there is some evidence supporting the idea. Another possibility is that dominant dRTA results from the association of mutant band 3 and wild-type band 3 into heterooligomers in the kidney, but not in erythroid cells, leading to intracellular retention or mistargeting of both proteins in the kidney and normal expression in red cells.
Carbonic anhydrase II (CAII) binds to the C-terminus of band 3 and aids in chloride-bicarbonate transport. Patients who lack CAII develop dRTA, osteopetrosis, and neurologic disease. This raises the possibility that some C-terminal band 3 dRTA mutations (see Fig. 16-10 ) may cause dRTA through loss of CAII function instead of, or in addition to, mistrafficking of band 3.
Protein 4.2 Defects: Recessive Hereditary Spherocytosis
HS caused by protein 4.2 deficiency is most common in Japan, although a few patients have been described in other populations. Overall, protein 4.2 mutations account for 9% of the published HS molecular defects. The characteristics of this form of HS are summarized in Table 16-8 .
| <5% of hereditary spherocytosis in the United States and Europe; 45%-50% of hereditary spherocytosis in Japan |
| Autosomal recessive |
| 95%-100% deficiency of protein 4.2, decreased amount of CD47 |
| 4.2 Nippon is common in Japan |
| Blood smear with variable red cell morphology: spherocytes, acanthocytes, ovalostomatocytes, normal discocytes. Spherocytes may not be the predominant morphology |
Multiple protein 4.2 mutations have been described ( Fig. 16-11 ). The most common one is 4.2 Nippon , in which the red cells contain only a small quantity of a 74/72-kD doublet of protein 4.2 instead of the usual abundant 72-kD species, because of an Ala142→Thr mutation that affects the processing of the protein 4.2 mRNA. Protein 4.2 Nippon –deficient membranes lose 70% of their ankyrin with low ionic strength extraction, and the ankyrin loss is blocked by preincubation of the membranes with purified 4.2, which suggests that protein 4.2 stabilizes ankyrin on the membrane. This hypothesis is supported by the observations that the amount of protein 4.2 is low in the red cell membranes of ankyrin-deficient nb/nb mice and in HS patients who lack one ankyrin gene. In addition, red cells from patients homozygous for 4.2 Nippon are fragile and have heat-sensitive skeletons, clumped intramembranous particles, and increased lateral mobility of band 3. A weakening of ankyrin–band 3 binding due to loss of protein 4.2 has also been observed. However, ankyrin lability is not evident in all patients with protein 4.2 deficiency. Patients whose red cells lack protein 4.2 also lack CD47. CD47 connects the band3–ankyrin–4.2 complex to the Rh complex of which CD47 is a part. Loss of CD47 may also be important because CD47 is recognized by an inhibitory receptor on macrophages that blocks phagocytosis.
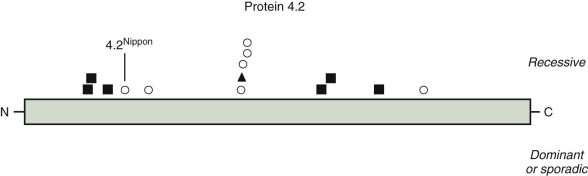
Finally, partial erythrocyte protein 4.2 deficiency has also been associated with mutations of band 3. Presumably, these mutations affect sites of band 3–protein 4.2 interactions.
Clinical Features.
Clinically, patients with HS due to protein 4.2 deficiency have mild to moderate HS. The peripheral smear sometimes contains typical spherocytes, but it may also contain ovalocytes, stomatocytes, or spiculated cells and relatively few spherocytes (see Fig. 16-7, E ). For this reason the diagnosis of HS may be missed in patients with protein 4.2 deficiency. The mechanism behind this unique and variable red cell morphologic picture is currently unknown.
Pathophysiology
Loss of Membrane Surface by Vesiculation
The primary membrane lesions in HS all involve vertical interactions between the skeleton and the bilayer and fit the theory that HS is caused by local disconnection of the skeleton from the bilayer and its integral membrane proteins, followed by vesiculation of the unsupported surface components. This, in turn, leads to progressive reduction in membrane surface area and to a shape called a spherocyte, although it usually ranges between a thickened discocyte and a spherostomatocyte.
Biomechanical measurements show that HS membranes are fragile. The force required to fragment the membrane is diminished and is proportional to the density of spectrin on the membrane. Membrane elasticity and bending stiffness are also reduced and are proportional to spectrin density. In addition, HS red cells lose membrane more readily than normal red cells when metabolically deprived or when their ghosts are subjected to conditions facilitating vesiculation. This has not been shown to occur in metabolically healthy HS spherocytes, perhaps because it occurs slowly (1% to 2% per day) under such conditions. However, massive vesiculation is evident in mice and zebrafish with severe spherocytic hemolytic anemias ( Fig. 16-12 ).
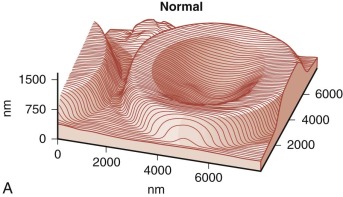
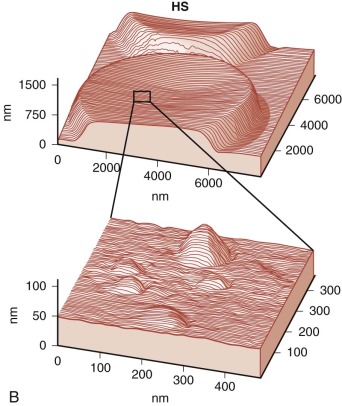
Because budding red cells are rarely observed in typical blood smears from patients with HS, the postulated vesiculation may either involve microscopic vesicles or occur in the reticuloendothelial system. Vesiculation may also occur in the bone marrow of patients with HS because similar decreases in the red cell surface area have been found in HS reticulocytes and in mature red blood cells. When membrane vesicles are induced in normal red cells, they originate at the tips of spicules, where the lipid bilayer uncouples from the underlying skeleton (see Fig. 15-29 ). The vesicles are small (≈100 nm) and devoid of hemoglobin and skeletal proteins and would be invisible by light microscopy. Tiny (50- to 80-nm) bumps have been detected by atomic force microscopy, on the surface of red cells obtained from patients with HS whose red cells are actively hemolyzing ( Fig. 16-13 ). The bumps could be microvesicles, although this needs to be proven. They are less than the length of a spectrin molecule (100 nm) and are not present on red cells from splenectomized patients.
Hypothesis 1.
The observation that spectrin or spectrin/ankyrin deficiencies are common in HS has led to the suggestion that they are the primary cause of spherocytosis. According to this hypothesis ( Fig. 16-14 , hypothesis 1), interactions of spectrin with bilayer lipids or proteins are required to stabilize the membrane. Spectrin-deficient areas would tend to bud off, leading to spherocytosis. However, this conjecture does not explain how spherocytes develop in patients whose red cells are deficient in band 3 but have normal amounts of spectrin.
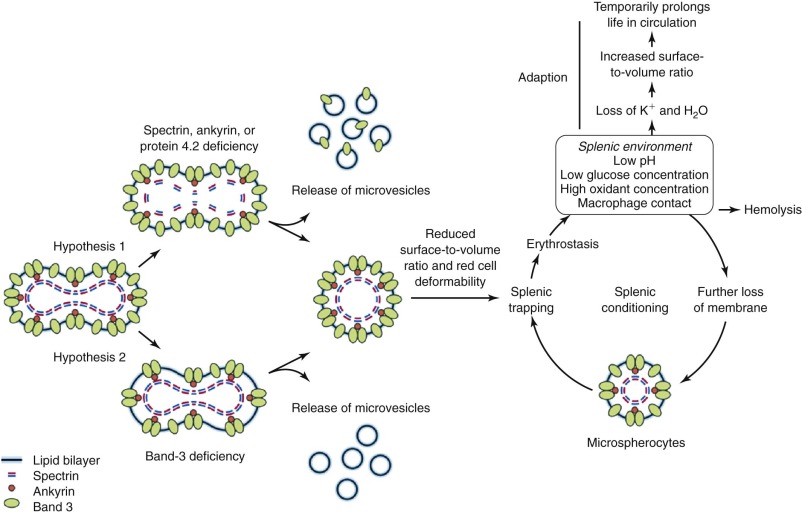
Hypothesis 2.
An alternate hypothesis argues that the bilayer is stabilized by interactions between lipids and the abundant band 3 molecules (see Fig. 16-14 , hypothesis 2). Each band 3 molecule contains about 12 hydrophobic transmembrane helices, many of which must interact with lipids. These interactions presumably spread beyond the first layer of lipids and influence the mobility of lipids in successive layers. In deficient red cells the area between band 3 molecules would increase, on average, and the stabilizing effect would diminish. Transient fluctuations in the local density of band 3 could aggravate this situation and allow unsupported lipids to be lost, resulting in spherocytosis. This hypothesis is supported by targeted disruption of the band 3 gene in mice. Erythrocytes from these mice lose massive amounts of membrane surface despite a normal membrane skeleton. This concept is also consistent with early studies of intramembrane particle aggregation, which leads to particle-free domains, as discussed in Chapter 15 . These domains are unstable, giving rise to surface blebs that are subsequently released from the cells as vesicles.
It is most likely that both of these hypotheses operate to a different degree depending on the membrane skeletal protein which is deficient—hypothesis 1 dominating in spectrin and ankyrin disorders and perhaps in protein 4.2 deficiency, and hypothesis 2 dominating in band 3 defects. Hypothesis 1 predicts that released microvesicles will contain band 3 and other skeleton-linked integral membrane proteins; hypothesis 2 predicts such vesicles will contain mostly lipids. This should be testable but has not yet been examined.
Loss of Cellular Deformability
Hereditary spherocytes hemolyze because of the rheologic consequences of their decreased surface-to-volume ratio. The red cell membrane is very flexible, but it can only expand its surface area about 3% before rupturing. Thus erythrocytes become less and less deformable as surface area is lost. Clever experiments show that the spleen begins to sense and restrain red cells that have lost as little as 2% to 3% of their membrane surface and that loss of more than 18% of the red cell surface leads to complete splenic sequestration. This corresponds to the 15% to 20% loss of membrane lipids and, presumably, membrane surface in HS red cells. For HS red cells, poor deformability is only a hindrance in the spleen, because the cells have a nearly normal life span after splenectomy.
Splenic Sequestration and Conditioning
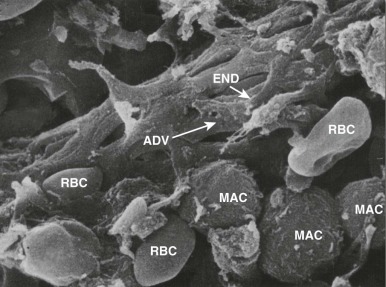
In the spleen most of the arterial blood empties directly into the splenic cords, a tortuous maze of interconnecting narrow passages formed by reticular cells and lined with phagocytes (see discussion of splenic structure and function in Chapter 23 ). Histologically, this is an “open” circulation, but most of the blood that enters the cords normally travels by fairly direct (i.e., functionally closed) pathways. If passage through these channels is impeded, red cells are diverted deeper into the labyrinthine portions of the cords, where blood flow is slow and the cells may be detained for minutes to hours. Whichever route is taken, to reenter the circulation, red cells must squeeze through spaces between the endothelial cells that form the walls of the venous sinuses ( Fig. 16-15 ). Even when maximally distended, these narrow slits are always much smaller than red cells, which are greatly distorted during their passage. Experiments have shown that spherocytes are selectively sequestered at this cordal-sinus juncture. As a consequence, spleens from patients with HS have massively congested cords and relatively empty sinuses. In electron micrographs, few spherocytes are seen in transit through the sinus wall, which contrasts to the situation in normal spleens, where cells in transit are readily found.
During detention in the spleen, HS red cells undergo additional damage marked by further loss of surface area and an increase in cell density. Many of these “conditioned” red cells escape the hostile environment of the spleen and reenter the circulation. In unsplenectomized patients with HS, two populations of spherocytes are detectable: a minor population of hyperchromic “microspherocytes” that form the “tail” of very fragile cells in the unincubated osmotic fragility test and a major population that may be only slightly more spheroidal than normal.
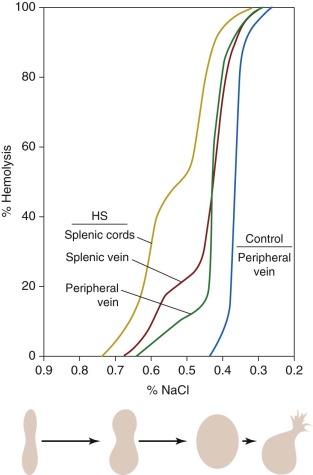
By 1913, it was known that HS red cells obtained from the splenic vein were more osmotically fragile than those in the peripheral circulation. However, the significance of this finding was not clear until the classic studies in the 1950s by Emerson and Young, who showed that osmotically fragile microspherocytes are concentrated in and emanate from the splenic pulp ( Fig. 16-16 ). After splenectomy, spherocytosis persists, but the tail of hyperfragile red cells is no longer evident on osmotic fragility testing. These and other data led to the conclusion that the spleen detains and “conditions” circulating HS red cells in a way that increases their spheroidicity and hastens their demise. The kinetics of this process were beautifully illustrated in vivo by Griggs, who found that a cohort of 59 Fe-labeled HS red cells gradually shifted from the major, less fragile population to the more fragile, conditioned population 7 to 11 days after entering the circulation. Although most conditioned HS red cells that escape the spleen are probably recaptured and destroyed, the damage incurred is sufficient to permit extrasplenic recognition and removal, because conditioned spherocytes, isolated from the spleen and reinfused postoperatively, are rapidly eliminated.
The mechanism of splenic conditioning is less certain. It is difficult to obtain accurate information about the cordal environment, but the existing data suggest it is metabolically inhospitable. Crowded red cells must compete for limited supplies of glucose in acidic surroundings (pH 6.8 to 7.2) where glycolysis is inhibited. The acidic environment induces Cl − and water entry and cell swelling but also stimulates the K + ,Cl − cotransporter, which produces a net loss of K + and water from the cells. The adverse effects of the cordal environment are further compounded by the presence of oxidant-producing macrophages. It has also been suggested that methylation of erythrocyte membrane proteins could contribute to the splenic conditioning of spectrin-deficient HS red cells. Hence, the spherocyte, detained in the splenic cords because of its surface deficiency, is stressed by erythrostasis in a metabolically threatening environment.
Erythrostasis
The spherocyte is particularly vulnerable to erythrostasis. This is the basis of the well-known autohemolysis test. During prolonged sterile incubation in the absence of supplemental glucose, red cells undergo a series of changes that culminate in hemolysis. The sequence of the changes is the same for HS and normal red cells; however, because HS cells are abnormally leaky and bear unstable membranes, their degeneration is accelerated.
HS red cells are initially jeopardized, because their membrane permeability to Na + is mildly increased. Their propensity to accumulate Na + and water is normally balanced by increased Na + pumping; however, the increased dependence on glycolysis is detrimental in erythrostasis, where substrate is limited. HS red cells exhaust serum glucose and become ATP depleted more rapidly than normal red cells. As ATP levels fall, ATP-dependent Na + ,K + and Ca 2+ pumps fail, and the cells gain Na + and water and swell. Later, when the Ca 2+ -dependent K + (Gárdos) pathway is activated, K + loss predominates and the cells lose water and shrink. The Na + gain is accelerated in HS red cells but is insufficient by itself to induce hemolysis. However, HS red cells are doubly jeopardized. As noted earlier, they are inherently unstable and fragment excessively during metabolic depletion. Membrane lipids (and probably integral membrane proteins) are lost at more than twice the normal rate. At first this surface loss is balanced by cell dehydration, but eventually (within 30 to 48 hours) membrane loss predominates, the cells exceed their critical hemolytic volume, and autohemolysis ensues.
Consequences of Splenic Trapping
Calculations indicate that the average normal red cell passes through the splenic cords about 14,000 times during its lifetime and has an average transit time of 30 to 40 seconds, surprisingly close to measured transient times in normal human spleens in vivo. The calculated residence time of the average HS red cell in the splenic cords is much longer, perhaps as long as 15 to 150 minutes, but still far short of the time required for metabolic depletion to occur. This conclusion is supported by direct analysis of splenic red cells. HS red cells obtained from the splenic pulp and containing 80% to 100% conditioned cells are moderately cation depleted and show changes in adenosine diphosphate (ADP) and 2,3-DPG concentrations consistent with metabolism in an acidic environment, but their ATP levels are normal. Others have reported similar results.
The data suggest that splenic conditioning is caused by mechanisms other than ATP depletion. For example, K + loss and membrane instability may be exacerbated by the high concentrations of acids and oxidants that must exist in a spleen filled with activated macrophages lunching on trapped HS red cells. In vitro, oxidants from activated phagocytes can diffuse across the membranes of bystander red cells and damage intracellular proteins within minutes. Red cells moving through the rapid transit pathways in the spleen might escape damage, but those caught in cordal traffic would be vulnerable. Oxidants, even in relatively low concentrations, cause selective K + loss by a variety of mechanisms and also damage membrane skeletal proteins. Finally, there is some evidence that HS red cells may be abnormally sensitive to oxidants. When exposed to peroxides they undergo remarkable blebbing and, presumably, vesiculation. If a similar process occurs in the spleen, it could be responsible for the excessive surface loss observed in conditioned cells.
The possibility that macrophages may directly condition HS red cells should also be considered. It is well known that spherocytosis often results from the interaction of immunoglobulin G (IgG)–coated red cells with macrophages, but HS red cells do not have abnormal levels of surface IgG. Macrophages also bear receptors for oxidized lipids (scavenger receptor) and phosphatidylserine, but there is no evidence at present that HS red cells expose the relevant ligands. The involvement of macrophages is supported by observations that large doses of corticosteroids markedly ameliorate HS in unsplenectomized patients. The effects are similar to those produced by splenectomy. It is well known that similar doses of corticosteroids inhibit splenic processing and destruction of IgG- or C3b-coated red cells in patients with immunohemolytic anemias, probably by suppressing macrophage-induced red cell sphering and phagocytosis. Electron microscopy shows that splenic erythrophagocytosis is common in HS, particularly in the splenic cords. In addition, phagocytes expressed from the cords of patients with HS contain bits of ghostlike “debris,” presumably resulting from membrane fragmentation.
Adaption: Benefits of Red Cell Dehydration for the HS Red Cell
It has been known for many years that HS red cells intrinsically leak more Na + and K + ions than normal cells. All the Na + transmembrane movements are increased, including passive diffusion, Na + ,K + -ATPase pump, Na + ,K + ,2Cl − cotransport, and Na + , Li + countertransport. The excessive Na + influx activates the Na + ,K + -ATPase pump, and the accelerated pumping, in turn, increases ATP turnover and glycolysis. At one time, it was believed that this modest Na + leak was responsible for the hemolysis of hereditary spherocytes, particularly those cells trapped in the unfavorable metabolic environment of the spleen, but it now is clear that this is incorrect, because the magnitude of the Na + flux does not correlate with the extent of hemolysis in HS.
The dehydration of HS red cells, as reflected in their high MCHC, is likely to be caused, at least in part, by the adverse environment of the spleen. This assumption has been made on finding that spherocytes from surgically removed spleens are the most dehydrated. The pathways causing HS red cell dehydration have not been clearly defined. One likely candidate is the Gárdos channel, a Ca 2+ -activated K + channel, which is known to contribute to erythrocyte dehydration in sickle cell anemia and thalassemia. One study showed that the Gárdos channel is functionally upregulated in a form of mouse HS caused by lack of erythrocyte protein 4.1R. When these mice were treated with clotrimazole, a known Gárdos channel blocker, hemolysis and anemia worsened, indicating that red cell dehydration protected the hereditary spherocytes, presumably by preserving their surface-to-volume ratio and therefore their flexibility in the face of surface loss.
Another possible cause of red cell dehydration in HS is increased K + ,Cl − cotransport, which is activated by acid pH. HS red cells, particularly from unsplenectomized subjects, have a low intracellular pH, reflecting the low pH of the splenic environment. The K + ,Cl − cotransport pathway is also activated by oxidation, which is likely to be caused by splenic macrophages.
Hyperactivity of the Na + ,K + pump, triggered by increased intracellular Na + , can dehydrate red cells directly, because three Na + ions are extruded in exchange for only two K + ions. The loss of monovalent cations is accompanied by water.
A fourth possible cause is the recently described mechanosensitive Piezo1 channel, which responds to membrane stretch and is thought to be involved in homeostatic adjustments to changes in volume. It is defective in hereditary xerocytosis, a disease characterized by red cell dehydration.
Whichever transport pathways cause HS red cells to lose K + and water, it is likely that the dehydration is a form of “adaption” ( Fig. 16-14 ) that prolongs the life of the HS red cell. How much it does so is unknown. Polymorphisms in cation channels like Piezo1 and calcium ATPase 4, in the globin loci, in major membrane skeletal proteins, and in genes like TAF3 , DNAJA4 , WDR61 and HBS1L-MYB are associated with variation in human MCHC. It will be interesting to see if these genes affect the severity of HS.
Laboratory Characteristics
Most of the classic laboratory features of HS are those found in other forms of hemolytic anemia, such as reticulocytosis, erythroid hyperplasia of the bone marrow, indirect hyperbilirubinemia, and increased fecal urobilinogens. The plasma hemoglobin level is often normal, and the haptoglobin value is only variably reduced, because most of the hemoglobin that is released when HS red cells are destroyed is catabolized at the site of destruction, so-called extravascular hemolysis. The spherocytic morphology combined with laboratory tests that measure a loss of surface area, such as the osmotic fragility test or eosin-5′-maleimide–binding test, distinguish HS from most other forms of hemolytic anemia. Membrane protein analysis and molecular genetic testing are only performed in research laboratories, as these tests are expensive and not very useful clinically. This is likely to change in the near future, though, as the cost of whole-exome and even whole-genome sequencing is falling very rapidly.
Red Cell Morphology
Spherocytes (see Fig. 16-2 ) are the hallmark of HS. They are dense, round, and hyperchromic; lack central pallor; and have a decreased mean cell diameter. They are always evident in blood smears from patients with moderate or moderately severe HS but are obvious in only 25% to 35% of patients with mild HS. Hereditary spherocytes are technically misnamed, as they range in shape from thickened discocytes to spherostomatocytes when examined under the scanning electron microscope.
In patients with HS, spherocytes and microspherocytes are the predominant abnormal cells on the peripheral blood smear (other than polychromatophils). However, there are morphologic variations that occur with some of the specific membrane defects in HS (see Fig. 16-7 ). Patients with ankyrin defects, the most common subgroup, have typical smears (see Fig. 16-7, C ), but many patients with β-spectrin defects also have a subpopulation of acanthocytes or hyperchromic echinocytes (see Fig. 16-7, B ) and most patients with band 3–deficient HS also have a small number of button mushroom–shaped red cells on their smears (see Fig. 16-7, D ). In severe HS, caused by homozygous or compound heterozygous α-spectrin mutations or by severe ankyrin defects, misshapen spherocytes, spiculated red cells, and bizarre poikilocytes may be seen and even dominate the blood smear (see Fig. 16-7, A ). Red cell morphologic findings are variable in protein 4.2 deficiency. Some patients have typical spherocytosis, but acanthocytes, echinocytes, ovalostomatocytes, and other poikilocytes have been observed in others (see Fig. 16-7, E ). Finally, some patients with truncated β-spectrin chains have spherocytic elliptocytosis (see later).
In contrast to red cells in IgG-mediated autoimmune hemolytic anemias, where spherocytosis also dominates the blood picture, HS red cells lose most of their surface area in the bone marrow instead of in the circulation. Nucleated red cells are uncommon in blood smears, except in the most severe forms of HS. Howell-Jolly bodies are also uncommon before splenectomy (4% of patients) and suggest reticuloendothelial blockade.
Red Cell Indices
Most patients have mild to moderate HS with mild anemia (hemoglobin level of 9 to 12 g/dL) or no anemia at all (so-called compensated hemolysis). In moderate to severe HS, the hemoglobin concentration ranges from 6 to 9 g/dL. In patients with the most severe disease, the hemoglobin concentration may drop to as low as 4 to 5 g/dL. The reticulocyte count is elevated without a comparable increase in the immature reticulocyte fraction.
The MCHC of HS red cells is increased due to relative cellular dehydration. The average MCHC exceeds the upper limit of normal (36%) in about one half of patients with HS, but all patients have some dehydrated cells. The Technicon H1 blood counter and its successors, which use the principle of flow cytometry (i.e., dual-angle laser light scattering), measure the mean corpuscular volume (MCV) and MCHC directly and provide histograms of both parameters. There is a right shift of the MCHC histogram in HS with an increased (>4%) population of hyperdense cells (MCHC > 41 g/dL) due to red cell dehydration, and a broadening of the distribution of MCVs, due to the mixture of microspherocytes and reticulocytes ( Fig. 16-17 ). This excess proportion of hyperdense cells is accurate enough to identify nearly all patients with HS and is one of the easiest and most accurate ways to diagnose the disease when one member of a family is already known to have HS.
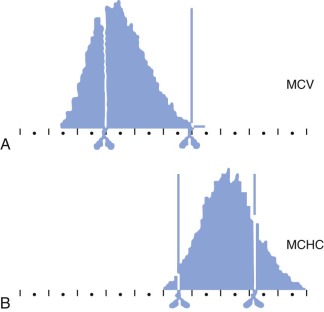
The MCV usually falls within the lower normal range in HS, but it is low in about 8% of adults and 16% of children, especially those with more severe HS. It is rare for the MCV to be less than 70 fL unless there is concomitant thalassemia or iron deficiency. However, the MCV is low relative to the age of the cells (reticulocytes have a high MCV) in all patients with HS, reflecting the dehydrated state of HS red cells. The reticulocyte MCV is also low, which contrasts with IgG-mediated autoimmune hemolytic anemia, where spherocytes are present but the reticulocyte indices are normal.
The RDW of HS erythrocytes is also very high, particularly in patients with more severe disease (see Table 16-3 ).
Various studies show that the MCHC or the percentage of hyperdense cells and the high RDW discriminate HS patients from normal individuals, though less so following splenectomy. An MCHC greater than 35 g/dL has a sensitivity of 70% and a specificity of 86% in diagnosing HS; an RDW greater than 14 has 85% sensitivity and 97% specificity. Combining the MCHC with the erythrocyte distribution width leads to a specificity approaching 100%. The RDW, MCHC, hemoglobin : MCHC ratio, and hemoglobin : RDW ratio all correlate with the severity of disease (see Table 16-3 ).
Fragility and Autohemolysis Tests
Osmotic Fragility Test.
Osmotic fragility testing (see Fig. 16-16 ) is performed by suspending red cells in increasingly hypotonic buffered NaCl. In hypotonic solutions, normal erythrocytes increase their volume by swelling until they become spherical and burst, releasing hemoglobin into the supernatant. Cells that begin with a decreased surface-to-volume ratio, like spherocytes or stomatocytes, reach the spherical limit at a higher NaCl concentration than normal cells and are termed osmotically fragile. Freshly drawn red cells from approximately one fourth of individuals with HS will have a normal or nearly normal osmotic fragility curve, approximating the number of HS patients with few spherocytes seen on their peripheral blood smears. However, after incubation at 37°C for 24 hours, HS red cells lose membrane surface area more readily than normal red cells, because their membranes are leaky and unstable. This incubation accentuates the defect in HS erythrocytes and brings out the defect of osmotic fragility. When the spleen is present, a subpopulation of very fragile erythrocytes that have been conditioned by the spleen form the “tail” of the osmotic fragility curve. This tail disappears after splenectomy.
The osmotic fragility test detects as abnormal (i.e., osmotically sensitive) any red cells with a decreased surface-to-volume ratio. This includes red cells in hereditary stomatocytosis (HSt), hereditary pyropoikilocytosis (HPP), autoimmune hemolytic anemia, and, sometimes, congenital dyserythropoietic anemia, type II (CDAII). Red cells with a large surface-to-volume ratio, whether owing to an increase in membrane surface, like target cells, or to cellular dehydration, like hereditary xerocytes, have decreased osmotic fragility (i.e., are osmotically resistant).
The incubated osmotic fragility test is thought to be more sensitive than the unincubated osmotic fragility test, although this is not well proven. For a long time the osmotic fragility test, particularly the incubated test, was the gold standard test for diagnosing HS, but recent comparisons show that other tests are more sensitive and specific.
Acidified Glycerol Lysis Test and Pink Test.
The glycerol lysis test was developed as an alternative to the osmotic fragility test and is widely used in Europe. In this test, red cells are incubated in glycerol-sodium phosphate–buffered hypotonic saline solution. The glycerol slows the entry of water into the cells, so that the time for the cells to lyse is prolonged and can be measured accurately. The glycerol lysis time is shortened for HS red cells because of the reduced surface-to-volume ratio.
The original glycerol lysis test lacked sensitivity and specificity, but the acidified glycerol lysis test (AGLT), and especially its incubated variant, have a high sensitivity (82% to 98%) and specificity (91% to 95%). Another variation achieves 100% sensitivity and specificity by changing the concentration of the sodium phosphate buffer from 5.3 to 9.3 mmol/L. This eliminates the need for preincubation. The modified test is easy to perform and requires very little blood, though accurate control of pH is critical. The simplicity of this test allows it to be used for rapid screening of a large number of blood samples.
Another adaptation of the original glycerol lysis test is the Pink test . Modifications of the test have made it adaptable to microsize samples (e.g., fingerstick blood samples). However, direct comparisons suggest that it is less specific than the osmotic fragility test or incubated AGLT.
The AGLT, like the osmotic fragility test, is often abnormal in patients with autoimmune hemolytic anemia, but it is also sometimes abnormal in a variety of other conditions, including CDAII, paroxysmal nocturnal hemoglobinuria, hereditary elliptocytosis, myelodysplastic syndromes, mechanical hemolysis, and chronic renal failure, and in pregnancy.
Hypertonic Cryohemolysis Test.
This method is based on the fact that HS red cells are particularly sensitive to cooling at 0°C in hypertonic solutions, which causes the cells to lyse and release hemoglobin. The molecular basis for this sensitivity is not understood, which is a disadvantage, but the test is reported to have a high sensitivity (90% to 100%) with one exception (79%), and to have good specificity (94% to 100%). It is very easy to perform, requires very little blood (a capillary blood sample is sufficient ), and is compatible with the common blood specimens anticoagulated with ethylenediaminetetraacetic acid (EDTA). These qualities make it a good test for large-scale screening. The hypertonic cryohemolysis test is not widely used but deserves more attention.
Autohemolysis Test.
The autohemolysis of erythrocytes after 48 hours at 37°C is normally less than 5% in the absence of glucose or less than 1% in the presence of glucose. Autohemolysis of spherocytes increases to 15% to 45% in the absence of glucose. In HS, the degree of autohemolysis is reduced by the addition of glucose, whereas in red cell glycolytic disorders and acquired disorders such as immune-mediated anemias, the degree of autohemolysis is not reduced. Although this differentiation may be occasionally helpful, the autohemolysis test is time consuming and cumbersome; gives variable results, with a sensitivity of only 70% to 80% and a specificity of 80% to 90% ; and is rarely performed.
Eosin-5′-Maleimide–Binding Test
When red blood cells are treated with the fluorescent probe eosin-5′-maleimide (EMA), 80% of the fluorophore binds to Lys430 on the outside surface of band 3. The other 20% binds to three proteins in the Rh complex: the Rh blood group proteins, RhAG, and CD47. Thus the measurement of EMA fluorescence detects loss of membrane surface if that loss includes band 3 or the Rh complex. The EMA test is usually done with a flow cytometer so the fluorescence of individual cells is quantified. Because membrane vesicles containing band 3 are believed to be lost in HS due to spectrin and ankyrin deficiency (see Fig. 16-14 ) and CD47 and, perhaps, band 3 molecules are lost in HS due to protein 4.2 deficiency, the EMA test detects all the different forms of HS, not just HS due to primary band 3 deficiency. The test is both highly sensitive (92% to 100%) and highly specific (94% to 100%) for HS. The only exception is one study that reported only 70% specificity. The test requires very little blood, does not require preincubation, takes little time to perform, is not affected by storage of red cells for a week, and is probably the single best test for diagnosing HS at the present time. It is recommended by the General Haematology Task Force of the British Committee for Standards in Haematology, and when combined with the AGLT achieved 100% sensitivity in one recent comparison of tests for HS. In particular, the test is normal in autoimmune hemolytic anemias, which give false-positive results in the osmotic fragility test, AGLT, and cryohemolysis test. The EMA test is abnormal in disorders where membrane surface is decreased or band 3 is abnormal, such as HPP, SAO, cryohydrocytosis, congenital dyserythropoietic anemia, type II, and Rh null disease. However, these are all rare disorders and are fairly easy to distinguish by red cell morphologic findings and other criteria.
Osmotic Gradient Ektacytometry
The ektacytometer is a laser diffraction viscometer, a device that measures how much red cells are deformed by shear stress. The device can be used to measure membrane mechanical strength by determining the shear stress needed to fragment isolated red cell membranes, or it can be used to assess red cell surface area, surface-to-volume ratio, and cellular hydration by measuring red cell deformation as a function of osmolarity at a constant shear stress. The latter method is termed osmotic gradient ektacytometry and is the gold standard method for diagnosing HS ( Fig. 16-18 ). One study demonstrated a 100% detection rate of HS using osmotic gradient ektacytometry and only a 66% detection rate of HS using the osmotic fragility test in the same population. Unfortunately, only a few laboratories in the world are equipped with an ektacytometer and the instrument is no longer commercially available.
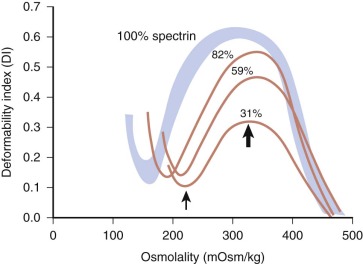
Membrane Protein, RNA, and DNA Measurements
Specialized tests are occasionally used to study patients with complicated disease or those for whom additional information is desired. Useful tests for these purposes include structural and functional studies of erythrocyte membrane proteins, such as protein quantitation, limited tryptic digestion of spectrin, membrane protein synthesis and assembly, and ion transport studies.
Membrane protein concentrations are usually assessed using SDS gels. Individual stained bands are quantified by densitometry or by eluting the dye from an excised band and measuring its concentration spectrophotometrically. This technique is satisfactory for detecting spectrin deficiency (expressed as a spectrin-to–band 3 ratio), although it is not as accurate as a radioimmunoassay or enzyme-linked immunoassay. With densitometry, spectrin and ankyrin deficiencies may be underestimated because they are normalized to band 3, which is partially lost along with membrane lipids as spherocytes circulate. As a result, the spectrin-to–band 3 ratio is lower after splenectomy. This is even more true for ankyrin, which is present in smaller amounts, migrates close to β-spectrin on SDS gels, and is elevated in reticulocytes. It is likely that patients with ankyrin defects, who have combined spectrin and ankyrin deficiency, are often misdiagnosed as having pure spectrin deficiency on SDS gels. SDS gels are satisfactory for detecting band 3 deficiency (elevated spectrin-to–band 3 ratio) or protein 4.2 deficiency, and they are useful in combination with antibody staining (Western blots) for detecting mutant proteins of altered size.
Moreover, SDS gels do not always identify the abnormal protein in HS, because missense mutations may cause functional defects rather than quantitative defects and because protein deficiencies may be as small as 10%. Therefore several other approaches have been described in an attempt to define specific molecular defects. In families with multiple affected members, linkage analysis has been performed to include or exclude the candidate genes. In addition, suspected proteins can be sequenced using direct nucleotide sequence analysis of polymerase chain reaction (PCR)–amplified complementary DNA (cDNA) or genomic DNA. Currently, no commercial laboratories identify the molecular defects in HS membrane proteins, and these analyses are performed only by a few research laboratories. However, with the rapidly falling costs of both whole-exome and whole-genome sequencing it is likely that DNA sequencing may become the most cost-effective way to diagnose HS (and many other inherited diseases) in the near future. Because HS membrane protein mutations are mostly restricted to specific families and do not predict the severity of the disease, in most cases it is not clinically important to know the exact molecular defect. More detailed studies, sometimes including DNA sequencing, should be considered in patients where the diagnosis is unclear after standard testing and in patients whose HS is more severe than in other family members, possibly due to coinheritance of another deleterious allele.
Differential Diagnosis
In general, HS is easily diagnosed if there are typical laboratory findings of spherocytosis, Coombs negative hemolysis, increased osmotic fragility, high MCHC, high RDW, and a positive family history. However, HS must be differentiated from other causes of spherocytosis, and there are several situations in which diagnosis can be difficult. Other causes of spherocytosis are listed in Table 16-1 .
ABO Incompatibility in Neonates
In the neonatal period it may be challenging to differentiate HS from ABO incompatibility. Anemia, hyperbilirubinemia, circulating microspherocytes, and altered osmotic fragility are found in both conditions, and results of the direct Coombs test are sometimes negative in ABO incompatibility. However, in most infants with ABO incompatibility and significant hemolysis, anti-A (or anti-B) antibodies can be eluted from the red cells, and free anti-A (or anti-B) IgG antibodies can be detected in the infant’s serum. The combination of HS and ABO incompatibility, though rare, is particularly severe.
Immunohemolytic Anemias
Occasionally, patients with immunohemolytic anemias and spherocytosis also have so few antibody molecules attached to their red cells that results of the direct Coombs test are negative. In these rare cases, the differentiation of the disease from HS is possible only with the use of radioactive antiglobulin reagents, the so-called super–Coombs test.
Heinz Body Hemolytic Anemias
Spherocytosis is also seen in Heinz body hemolytic anemias during an acute hemolytic crisis and occasionally in the steady state. However, in this situation, bite cells and/or blister cells and various dense, irregular cells are always present on the peripheral blood smear, and Heinz bodies can often be detected in red cells supravitally stained with methyl violet.
Aplastic Crises
Diagnostic difficulties may also arise in HS patients who are seen during an aplastic crisis (see below). Early in the crisis, the acute nature of the symptoms may suggest an acquired process and the absence of reticulocytes may divert the physician from a diagnosis of hemolytic anemia. Later, as marrow function returns, physicians may occasionally be misled by the properties of the emerging young HS red cells, which are less spherocytic and osmotically fragile than usual.
Congenital Dyserythropoietic Anemia, Type II
In a preliminary report, Mariani et al noted that 13% of cases referred to their reference laboratory with a provisional diagnosis of HS were found to have CDAII, a disorder featuring underglycosylated band 3 and prominent binuclearity of late erythroblasts that is caused by defects in the secretory pathway gene SEC23B (see Chapter 7 ). Although CDAII patients typically display a variety of morphologic findings, a subset have prominent spherocytosis or elliptocytosis.
Hereditary Stomatocytosis and Xerocytosis
As discussed later in this chapter, patients with disorders of cation permeability, especially hereditary stomatocytosis (HSt) and hereditary xerocytosis (HX), may be misdiagnosed as having HS because they have a dominant hemolytic anemia with an abnormal osmotic fragility test. Care must be taken to exclude these disorders in any HS patient who is to be treated with splenectomy because there is a high risk of thromboembolic complications following splenectomy in patients with HSt and HX.
Conditions That Camouflage Hereditary Spherocytosis
HS may be camouflaged by disorders that increase the surface-to-volume ratio of the red cells, such as iron deficiency, vitamin B 12 or folate deficiency, obstructive jaundice, β-thalassemia, or sickle cell–hemoglobin C disease (hemoglobin SC disease). In these difficult cases, additional tools, such as red cell membrane protein analysis, or exome or genome sequencing may be necessary to confirm the diagnosis of HS.
Iron Deficiency
Iron deficiency corrects the abnormal shape, fragility, and high MCHC of hereditary spherocytes but does not improve their life span. Megaloblastic anemia can mask the morphologic characteristics of HS. Osmotic fragility is also improved. The masking effect is observed in both vitamin B 12 and folate deficiencies and is rapidly reversed after correction of the nutritional deficit.
Obstructive Liver Disease
When obstructive jaundice develops, spherocytosis and osmotic fragility transiently improve and hemolysis abates. This is due to the expansion of red cell membrane surface area that follows the increased uptake of cholesterol and phospholipids from the abnormal plasma lipoproteins that are released by the obstructed liver. In normal cells, this increase in surface area leads to the formation of target cells, but in spherocytes, it leads to the appearance of discocytes. For example, we have seen a 6-year-old child who developed jaundice and symptoms of biliary obstruction. She had a palpable spleen tip, evidence of mild compensated hemolysis (hemoglobin concentration of 14 g/dL; reticulocyte count of 3.3%), a normal peripheral blood smear, and normal results for an osmotic fragility test (fresh and incubated cells). Abdominal radiographic studies showed calcified stones in the gallbladder and common bile duct. After cholecystectomy and relief of the partial biliary obstruction, the child’s hemolysis worsened (reticulocyte count of 10.8%) and she developed anemia (hemoglobin concentration of 10.2 g/dL), spherocytosis, and abnormal results on an incubated osmotic fragility test. She subsequently underwent splenectomy with clinical improvement.
β-Thalassemic Trait
The coexistence of HS and β-thalassemia trait has been described in a few case reports and has been reported to worsen, ameliorate, or have no effect on the clinical status of the patient. In a large French family with independently segregating HS and β-thalassemia trait, patients with both traits had signs of both diseases: small, hemoglobin A 2 -rich, osmotically fragile cells and some spherocytes on peripheral blood smears. However, the HS phenotype in these patients was less severe than that of family members with HS but without β-thalassemia trait.
Hemoglobin SC
Coinheritance of HS and hemoglobin SC disease may exacerbate the hemoglobinopathy. A 15-year-old male with both diseases was much more anemic than his siblings with hemoglobin SC disease and experienced five splenic sequestration crises. However, the two diseases may also disguise each other morphologically. Only a few spherocytes or target cells were evident on the boy’s blood smear, and the surface-to-volume ratio of his red cells (and probably their osmotic fragility) was normal. This is likely due to the balancing effects of HS (loss of surface area) and hemoglobin SC (loss of cell volume) on red cells.
Hemoglobin AS
Sickle trait may also be worsened by HS. Yang and colleagues reported two patients with the combination who suffered multiple splenic sequestration crises. Splenic infarcts also occur. On the other hand, spontaneous regression of HS, presumably due to development of a hyposplenic state, has also been observed in two family members who had HS and sickle cell trait.
Complications
Most patients with HS have reasonably well-compensated hemolysis and are rarely symptomatic. These patients only seek medical attention when complications occur. Complications are generally related to chronic hemolysis and anemia.
Gallstones
The formation of bilirubinate gallstones is one of the most common complications of HS and is a major impetus for splenectomy in many patients. Instances of gallstones occurring in infancy have been reported, but most appear in adolescents and young adults. In a retrospective study of 152 consecutive patients with HS, conducted before the development of ultrasonography, gallstones were detected in only 5% of children younger than age 10 years who were adequately examined and in approximately 50% of HS patients between 10 and 30 years of age ( Fig. 16-19 ). More recent data are similar; about 15% of patients less than 18 years old develop gallstones. The increase in the incidence of gallstones after age 30 years parallels the incidence in the general population, which suggests that cholelithiasis owing to HS is primarily manifested in the second and third decades.
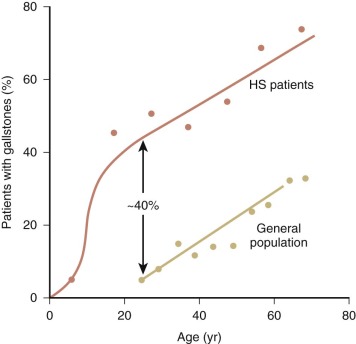
The incidence of gallstones in HS patients is related to the ability of the liver to metabolize increased amounts of bilirubin. A common polymorphism in the promoter of the uridine diphosphate-glucuronyl transferase gene ( UGT-1A ) causes decreased enzyme production and, in the homozygous form, causes Gilbert syndrome. Coinheritance of Gilbert syndrome with HS increases the risk for cholelithiasis. One retrospective study of 103 children with HS showed that the rate of gallstone formation was 2.2 times higher in those patients with a homozygous mutation in the UGT-1A gene than in those with a heterozygous mutation and 4.5 times higher than in healthy patients. HS patients with a homozygous mutation in the UGT-1A gene also develop gallstones at a younger age. Therefore analysis of this allele is useful in predicting the risk of cholelithiasis in HS.
Because the pigment stones typical of HS are easily detected by ultrasonography, all patients with HS should have ultrasound examinations about every 5 years if the spleen is intact and just prior to splenectomy. Unfortunately, longitudinal prospective studies using modern techniques (i.e., liver and biliary tree ultrasonography) are not available, making the true incidence and natural history of gallstones in this population unknown. One recent retrospective study showed that patients with HS (or other hemolytic anemias) were much more likely to develop symptomatic disease (i.e., pain, emesis, obstructive jaundice) than children with gallstones for other reasons. In that study, 68% of HS patients with gallstones had symptoms and all of them required cholecystectomy. This coincides with earlier reports. The most common complication was biliary obstruction, followed by acute cholecystitis. However, studies that include large numbers of patients with mild HS show a much lower incidence of symptomatic gallbladder disease.
More accurate, longitudinal studies are needed to determine the optimal treatment of patients with mild to moderate HS and cholelithiasis, particularly asymptomatic cholelithiasis that is detected on routine screening. Only about 15% to 20% of adults with asymptomatic stones (all types) develop symptoms over a prolonged period, but the retrospective data above suggest the complication rate may be much higher for bilirubinate stones. Surgery is indicated for recurrent symptoms or signs of biliary obstruction. It is unclear whether children who are undergoing cholecystectomy for symptomatic cholelithiasis should also have a splenectomy or partial splenectomy if it is not otherwise indicated (see later). Most authorities recommend it, arguing that the risk of stones persists, presumably in the remaining biliary radicals or cystic duct stump. However, cholelithiasis after cholecystectomy for typical gallbladder disease is rare unless a gallbladder remnant remains. Presumably the risk would be higher in someone with continuing hemolysis, but it is not clear how much higher.
If an HS patient requires a splenectomy, an ultrasound study of the gallbladder is indicated prior to surgery to assess the need for concomitant cholecystectomy. Cholecystotomy (removal of stones but not the gallbladder) is an option in children with silent stones because the risk of new stones forming is markedly reduced after splenectomy. For this reason, prophylactic cholecystectomy at the time of splenectomy solely to prevent the formation of gallstones is not indicated.
Crises
Patients with HS, like patients with other hemolytic diseases such as sickle cell disease, face a number of potential crises. Table 16-9 summarizes the types of crises that occur in patients with HS.
| Type | Anemia | Reticulocytes | Jaundice | Cause | Comment |
|---|---|---|---|---|---|
| Hemolytic | Increased | Increased | Increased | Viral infections leading to splenomegaly | Frequent, usually mild |
| Aplastic | Increased | Decreased | Decreased | Parvovirus B19 | Occurs once. Severe. Risk to pregnant contacts |
| Megaloblastic | Increased | No change or decreased | No change or increased | Relative folic acid deficiency | Rare, preventable |
Hemolytic Crises.
Hemolytic crises are probably the most common type in HS. They usually occur with viral syndromes, particularly in children younger than 6 years of age, and are presumed to be due to activation of the reticuloendothelial system. They are characterized by anemia, vomiting, abdominal pain, an increase in reticulocytes, a mild transient increase in jaundice, and tender splenomegaly. Severe hemolytic crises are rare and medical intervention with hospitalization and transfusions is seldom required.
Aplastic Crises.
Aplastic crises are less common than mild hemolytic crises but are more serious and may lead to severe anemia, resulting in congestive heart failure or even death. They tend to occur in infants and children. Aplastic crises are caused by parvovirus B19, the etiologic agent in erythema infectiosum (fifth disease). Parvovirus B19 infection may present as fever, chills, lethargy, vomiting, diarrhea, and myalgias. Normal children may also develop a maculopapular rash on the face (slapped cheek appearance), trunk, and extremities—the rash of fifth disease—but this is less common in children with hemolysis and an aplastic crisis. In addition, patients with an aplastic crisis experience pallor and weakness from their worsening anemia. Jaundice, if present, may decrease as the number of abnormal red cells that can be destroyed declines.
Parvovirus B19 selectively infects erythropoietic precursors and inhibits their growth by inducing apoptosis and cell cycle arrest at G 2 . Parvovirus infections are often associated with mild neutropenia (≈20%) or thrombocytopenia (≈40%), and instances of transient pancytopenia have been reported. Parvovirus may infect several members of a family simultaneously.
The sequence of events in an aplastic crisis was well described by Owren (though he confusingly called the crisis ”hemolytic,” reflecting the recovery phase, instead of aplastic) and is illustrated in Figure 16-20 . During the aplastic phase, the hematocrit level and reticulocyte count fall, marrow erythroblasts disappear, and unused iron accumulates in the serum. As production of new red cells declines, the cells that remain age and microspherocytosis and osmotic fragility increase. The return of marrow function is heralded by a fall in the serum iron concentration and the emergence of granulocytes, platelets, and, finally, reticulocytes. During this recovery phase, the serum phosphorus can drop to dangerously low levels. Furthermore, the lack of reticulocytes early in the recovery phase should not eliminate hemolytic anemias from diagnostic consideration.
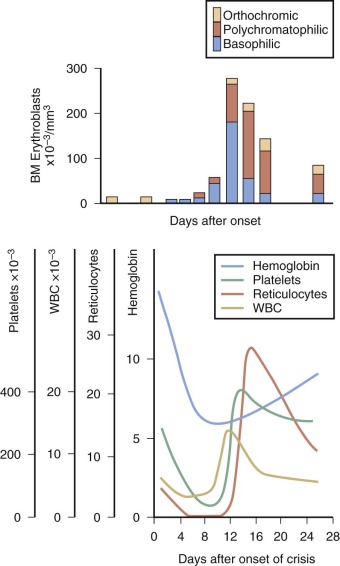
Infection with parvovirus B19 is a particular danger to susceptible pregnant women because it can infect the fetus, leading to fetal anemia, nonimmune hydrops fetalis, and fetal demise. The risk of nonimmune hydrops fetalis to the fetus of a woman who becomes infected with B19 during pregnancy is probably low. Nevertheless, because the virus is highly contagious and is easily transmitted to patients and staff and because only one half to two thirds of pregnant women have acquired protective antibodies, patients who have or are suspected of having an aplastic crisis should be placed under respiratory and contact precautions while hospitalized and IgG-negative contacts who are pregnant should be tested for evidence of seroconversion. Nonimmune hydrops fetalis can be detected by ultrasonography and treated with intrauterine transfusions.
Many asymptomatic patients with mild HS and compensated hemolysis first come to medical attention during an aplastic crisis. It is important to recognize that other family members with HS are susceptible and need to be tested. Diagnostic confusion may arise during reemergence of marrow function, when the physician may mistake an aplastic crisis for a hemolytic one. Because aplastic crises usually last for 10 to 14 days (about one half the life span of HS red cells), the hemoglobin value typically falls to about one half its usual level before recovery occurs. Thus aplastic crises are a serious threat to young children with HS, particularly children with more severe forms of the disease.
The diagnosis of parvovirus can be made using serum PCR or immunologic tests. Bone marrow pathology demonstrates giant pronormoblasts, which are a hallmark of the cytopathic effects of parvovirus B19. Treatment is supportive until the aplastic episode self-resolves. For severe anemia, red cell transfusions may be necessary. In immunocompromised patients, intravenous immunoglobulin assists in clearing the virus. Immunity after parvovirus infection is lifelong.
Megaloblastic Crises.
Rarely, HS patients may have megaloblastic crises due to folate deficiency. These crises occur in patients whose dietary intake of folic acid is inadequate for the increased needs of the red cell precursors in the bone marrow. Therefore megaloblastic crises typically occur in patients with HS who are recovering from an aplastic crisis or who are pregnant. A megaloblastic crisis in pregnancy has been reported as the first manifestation of HS. All patients with moderate to severe HS should routinely receive folic acid supplements (about 1 mg/day) prior to splenectomy to prevent this complication. Folate supplementation is unnecessary in mild HS or in HS following splenectomy except during pregnancy.
Other Complications
Gout and Leg Ulcers.
Rarely, adults with HS develop gout, indolent leg ulcers, or a chronic erythematous dermatitis on the legs. Splenectomy appears to be curative.
Extramedullary Hematopoiesis.
Adults with HS may also develop extramedullary masses of hematopoietic tissue, particularly alongside the posterior thoracic, lumbar, or sacral spine or in the hila of the kidneys or the adrenal gland. These masses may spontaneously bleed, leading to hemothorax. They may also cause spinal cord compression. Surprisingly, these tumors often arise in patients with mild HS, perhaps because they often do not undergo splenectomy, and may be the first manifestations of HS. The masses gradually enlarge and are sometimes mistaken for neoplasms. Biopsy may lead to extensive bleeding. If a biopsy is necessary, an open biopsy should be performed. These bone marrow tumors can be diagnosed by magnetic resonance imaging, which may make biopsies unnecessary. The masses stop growing and undergo fatty metamorphosis after splenectomy, but they do not shrink in size.
Hemochromatosis.
Iron overload is uncommon in HS. Only 4% of unsplenectomized, untransfused HS patients have a serum ferritin greater than 500 ng/mL. However, untreated HS may exacerbate hemochromatosis in patients who are heterozygous for the hereditary disease. Several patients with HS subsequently died of liver failure or hepatoma.
Angioid Streaks.
These brownish or gray streaks resembling veins in the optic fundus have been described in adult members of several HS kindreds. The rate of occurrence of this association is unknown. Angioid streaks are relatively common in some other hematologic disorders, notably sickle cell disease and thalassemia, and may be complicated by retinal vascular proliferation that requires treatment.
Pseudohyperkalemia.
Because HS red cells leak potassium ions more rapidly than normal red cells, serum samples from HS patients may demonstrate pseudohyperkalemia if allowed to sit for a long period of time before electrolytes are analyzed.
Splenic Rupture.
Although splenomegaly is a common symptom in HS patients, cases of splenic rupture are rare. In one large recent study, the risk of blunt splenic injury from trauma was only 1 in 20,000 to 50,000 per person per year, a rate that did not differ from the risk in the general population. This contrasts significantly with the splenomegaly that occurs during acute viral infections, such as Epstein-Barr virus, in which splenic rupture occurs with a higher frequency. It is unclear why this difference exists, although it may reflect the unique pathophysiology of splenic enlargement in the different conditions. Children with HS and splenomegaly should not be restricted from normal activities. However, older children and adults with splenic enlargement that extends below the rib cage should probably avoid activities that may inflict a powerful, direct blow to the abdomen.
Nonerythroid Manifestations.
Kindreds with HS and neurologic manifestations have been described. In the older literature, multiple case reports describe patients with HS and various neurologic abnormalities, such as a Friedrich ataxia-like disease, cerebellar disturbances, muscle atrophy, and a tabeslike syndrome. More recently, patients with certain ankyrin gene deletions have been reported, who have psychomotor retardation and various neurologic manifestations with or without hypogonadism. Additional reports describe patients with ankyrin-deficient HS and slowly progressive spinocerebellar disease. Erythroid ankyrin and β-spectrin are expressed in the cerebellum and spinal cord, and mice that lack ankyrin develop delayed cerebellar ataxia owing to a slow loss of Purkinje cells. This suggests that selected ankyrin and β-spectrin mutations may affect the neurologic system in addition to red blood cells.
A three-generation Russian family with co-segregating HS and hypertrophic cardiomyopathy has been described, and two brothers have HS, a movement disorder, and myopathy. These cases are interesting because the erythroid forms of spectrin and ankyrin are known to be expressed in muscle.
Splenectomy
Splenectomy cures all but the most severe cases of HS, eliminating anemia and hyperbilirubinemia and reducing the reticulocyte count to nearly normal levels (1% to 3%). In most patients, red cell survival becomes normal or remains only slightly shortened. Spherocytosis and altered osmotic fragility persist, but the “tail” of the osmotic fragility curve, created by conditioning of a subpopulation of spherocytes by the spleen, disappears. After splenectomy, patients with the most severe forms of HS still have some hemolysis, but their clinical improvement is striking. Splenectomy appears to have a more beneficial effect on the deformability of spectrin/ankyrin-deficient erythrocytes than band 3–deficient erythrocytes.
Currently, the major decisions about treatment are who should have a splenectomy, what kind of operation should be performed, and how patients should be cared for postoperatively. The indications for splenectomy have to be weighed carefully because a small fraction of patients will die of overwhelming postsplenectomy infections. Because the risk of postsplenectomy sepsis is very high in infancy and early childhood, splenectomy should be delayed until the age of 5 to 9 years if possible and to at least age 3 years in all children, even if transfusions are required chronically in the interim. There is no evidence that further delay is useful, and it may be harmful because the risk of cholelithiasis increases substantially after age 10 years (see Fig. 16-19 ).
Risks of Splenectomy
Immediate Postsplenectomy Complications.
Early complications of splenectomy include local infection such as a subphrenic abscess, bleeding, and pancreatitis due to intraoperative injury to the tail of the pancreas.
Postsplenectomy Sepsis.
Overwhelming serious bacterial infection with encapsulated bacterial organisms is one of the most significant risks of splenectomy. The risk is highest with encapsulated organisms, such as Streptococcus pneumoniae , Neisseria meningitidis , and Haemophilus influenzae . Postsplenectomy sepsis can be rapidly fatal, and it is unclear how the risk of postsplenectomy sepsis changes over time. Some studies have suggested that the risk is highest immediately after splenectomy, and decreases over time. Other studies conclude that the risk of sepsis is present indefinitely after splenectomy.
It is difficult to estimate the risk of postsplenectomy infections after infancy. The surveys of Schwartz and colleagues, Green and coworkers, and Schilling are limited to adults and largely predate immunization for S. pneumoniae and other bacteria. These surveys show an incidence of fulminant sepsis in adults of 0.2 to 0.5 per 100 person-years of follow-up and a death rate of about 0.1 per 100 person-years. Three of the four deaths in the Schilling study occurred 18 years or more after the operation, and none of the patients who died had received pneumococcal vaccine or prophylactic antibiotics. In addition, other serious bacterial infections (e.g., pneumonia, meningitis, peritonitis, and bacteremia) in adults were much more common (4.5 per 100 person-years) than normal, particularly in the first few years after the operation. The incidence of postsplenectomy sepsis appears to be higher in children, particularly younger children.
Unfortunately, the majority of studies of postsplenectomy sepsis in adults and children have serious methodologic problems. Most are case reports or retrospective reports subject to recall bias. The patients included in the studies have heterogeneous diseases and have often undergone splenectomy for a variety of indications. Furthermore, in most of the studies, the adult patients are not fully immunized and do not receive antibiotic prophylaxis. Further research is necessary to quantitate the risk over time of postsplenectomy sepsis in HS patients treated according to current practice guidelines.
With the development of the Haemophilus influenzae type B conjugate vaccine, meningococcal conjugate and nonconjugate vaccines, and pneumococcal conjugate and nonconjugate vaccines, the risk of postsplenectomy sepsis should be reduced. However, Grace found that only 26% of adults with HS who were splenectomized prior to 1990 had received all of these vaccines and most did not continue to take their antibiotic prophylaxis as adults. A second study came to similar conclusions. Few patients were adequately educated about antibiotic prophylaxis and fever management. These studies highlight the necessity for quality improvement in the care of HS patients after splenectomy.
Babesiosis and Malaria.
Splenectomized HS patients are at increased risk for serious parasitic infections, such as babesiosis and malaria. Babesiosis is a zoonotic infection in which an intraerythrocytic protozoan is transmitted by ticks; the vector is endemic in Europe and in the northeastern, upper Midwestern, and coastal northwestern United States. Babesiosis can also be transmitted by transfusion. About 20 cases per year are reported. In the United States, the etiologic agent of babesiosis is Babesia microti . The clinical symptoms usually begin with chills, anorexia, and fatigue and are followed by intermittent fever and symptoms including myalgias, arthralgias, headache, and vomiting. In healthy patients, the infection is often asymptomatic or mild. In patients with asplenia, severe illness with hemolytic anemia can occur, including fulminant illness resulting in death. Diagnosis is made through microscopic identification on Giemsa- or Wright-stained thick or thin blood smears. Serologic tests for Babesia antibodies and PCR are also available. Because the primary vector for the parasite is the Ixodes scapularis tick, which can also transmit diseases such as Lyme disease and granulocytic anaplasmosis (previously called granulocytic ehrlichiosis), these infections should be considered as well. Current treatment for severe babesiosis uses clindamycin and oral quinine for 7 to 10 days. Azithromycin and atovaquone are effective in milder disease. Exchange blood transfusions should be considered for severely ill patients, especially those with parasitemia of more than 10%. Splenectomized HS patients, when traveling in endemic areas, should attempt to avoid tick bites by wearing long pants and using tick repellants.
Animal experiments demonstrate that the spleen is essential in eliminating malaria parasitemia, and human observations show that in the absence of the spleen parasitemia is greater and more mature parasites (schizonts and trophozoites) are observed in the peripheral blood (reviewed ). However, only anecdotal reports or small series exist that demonstrate an increased risk of Plasmodium falciparum and Plasmodium vivax malaria in asplenic individuals, some of whom had unusually severe disease and others of whom did not. This may reflect the fact that in previously immune patients the spleen is almost nonessential for protection against severe malaria. We conclude that the risk of malaria in splenectomized individuals is not well documented. Nevertheless, the author knows experienced hematologists practicing in areas endemic for malaria who will not allow their patients to be splenectomized. This “local knowledge” suggests the risk is higher than the limited number of reports indicates. Accordingly, we believe that splenectomized HS patients should strictly adhere to malaria chemoprophylaxis when traveling in endemic areas or avoid such areas altogether, and should immediately seek medical advice if a fever develops.
Thrombosis and Thromboembolic Diseases.
Many studies demonstrate an increased risk of thrombosis in splenectomized patients where hemolysis persists after the operation, but it is not clear to what extent this risk applies to patients with HS. Major large-vessel and organ thrombosis is a common finding in murine models of severe HS, but thrombotic complications are rare in the milder forms of HS that are typical in humans, at least in the short term. Several studies from Schilling indicate that there is a risk in human HS as well, but it is not manifest for decades. Schilling has a unique experience, with up to five decades of follow-up of a cohort of 634 HS patients. His observations are summarized in Table 16-10 .
| Cumulative Incidence of First Event by Age 70 * | Hazard Ratio † | 95% Confidence Interval | P-value | |
|---|---|---|---|---|
| Arterial events ‡ | ||||
| Normals (male) | 17% | |||
| Normals (female) | 10% | |||
| Hereditary spherocytosis (HS) spleen out (male) | 32% | |||
| HS spleen out (female) | 22% | |||
| HS spleen in (male) | 2% | |||
| HS spleen in (female) | 3% | |||
| Male gender | 1.73 | 1.02-2.93 | 0.04 | |
| HS with spleen in versus normals | 0.22 | 0.08-0.58 | 0.002 | |
| HS with spleen out versus normals | 1.56 | 0.90-2.68 | 0.11 | |
| HS with spleen out versus HS with spleen in | 7.15 | 2.81-18.2 | <0.0001 | |
| Venous events § | ||||
| Normals (male) | 12% | |||
| Normals (female) | 4% | |||
| HS spleen out (male) | 19% | |||
| HS spleen out (female) | 20% | |||
| HS spleen in (male) | 4% | |||
| HS spleen in (female) | 7% | |||
| Male gender | 0.86 | 0.42-1.75 | 0.68 | |
| HS with spleen in versus normals | 0.90 | 0.26-7.13 | 0.87 | |
| HS with spleen out versus normals | 3.00 | 1.26-7.13 | 0.01 | |
| HS with spleen out versus HS with spleen in | 3.33 | 1.13-9.79 | 0.03 |
* Splenectomy prior to 18 years of age.
† From Cox proportional hazards regression model.
‡ Arterial events include myocardial infarction, stroke, coronary artery surgery, and carotid artery surgery.
§ Venous events include thrombophlebitis, deep vein thrombosis, and pulmonary embolism.
First, splenectomized HS patients have a significantly increased risk (threefold) of venous thrombotic events compared with normals. These included episodes of thrombophlebitis, deep vein thrombosis, and pulmonary embolism. There is also a modest (1.5-fold) but not quite statistically significantly increased risk of arterial events such as myocardial infarction, stroke, coronary artery disease requiring surgery, and carotid artery disease requiring surgery compared with normals. By age 70 years, 32% of men and 22% of women have experienced an arterial event and about 20% of each gender have had a venous event (see Table 16-10 ). These numbers are two to five times greater than in normal family members. In HS patients splenectomized in childhood or adolescence, venous thrombotic events begin to occur after age 30 years but arterial complications do not increase significantly until 55 to 60 years of age. The incidence of arterial events occurring following splenectomy shown in Table 16-10 is comparable to the 1.86-fold greater incidence of death from ischemic heart disease following splenectomy that Robinette and Fraumeni observed in an excellent case-controlled study of the outcomes of World War II battlefield splenectomies after three decades.
A few instances of pulmonary hypertension have been observed in HS patients years after splenectomy. Pulmonary hypertension is a known complication of splenectomy for other reasons, including nonhemolytic conditions such as Gaucher disease, but it was not unusually common in Schilling’s long-term study of HS patients, which corresponds to the recently reported experience of Crary et al and the unpublished experience of R.F. Grace at Boston Children’s Hospital. Portal vein thrombosis, priapism, and osteonecrosis also seem to be more prevalent after splenectomy for conditions such as thalassemia and Gaucher disease but do not seem to be a complication of splenectomy for HS.
The most interesting and surprising finding in Schilling’s studies is that HS patients who avoid splenectomy have about fivefold fewer arterial events than normals and sevenfold fewer events than HS patients who have had their spleens removed (see Table 16-10 ). This is probably because they have lower hemoglobin levels and higher bilirubin levels than normal. A low hemoglobin level is known to correlate with a lower risk of myocardial and cerebral infarction in the Framingham study. This may be because anemia of all types is associated with a low cholesterol level. This fact has been known for over 100 years but is little studied, is not understood, and has largely been forgotten. Whether the lower cholesterol level accounts for all of the vascular benefits of a low hemoglobin level is not known. Homocysteine concentrations rise after splenectomy for HS and may also be a contributing factor. Higher bilirubin levels also correlate with a low risk of coronary artery disease, perhaps because bilirubin is an antioxidant. It is possible that this benefit accounts for the prevalence of the Gilbert polymorphism.
Thus most of the sevenfold increased risk of deleterious arterial events following splenectomy in HS seems to result from relief of the anemia and hemolysis, whereas most of the threefold increased risk of venous thromboembolism seems to be due to the splenectomy itself (see Table 16-10 ). The factors potentiating thromboembolism are not well understood. Potential factors include (1) a chronically high platelet count, (2) decreased splenic clearance of free hemoglobin, which decreases serum nitric oxide and potentially causes vasocontriction, and (3) diminished splenic clearance of “blood dust” (microvesicles released from HS red cells and platelets). These particles are assumed to have phosphatidylserine exposed on their surfaces, thus activating the clotting cascade.
Although the vascular risks of splenectomy in HS observed by Schilling need to be confirmed by others, a conservative approach to splenectomy is supported by the available data. It is not clear to what extent polymorphisms that are known to raise the risk of hypercoagulation may contribute to the risks imposed by splenectomy, disorders such as factor V Leiden polymorphism, antithrombin III deficiency, deficiency of protein S or C, and the prothrombin G20210A mutation (see Chapter 33 ), but it is reasonable to assume they do contribute and incorporate them into the decision making as to whether a splenectomy is indicated or not. Further, once a patient with HS has been splenectomized attempts should be made to limit additional hypercoagulable risk factors.
Indications for Splenectomy
Splenectomy should only be performed if there are clear indications. All patients with severe spherocytosis, who are transfusion dependent or have growth failure or skeletal changes, should undergo splenectomy. Full or partial (subtotal) splenectomy (see later section) is also usually recommended for patients with moderate HS if they suffer from reduced vitality or physical stamina due to anemia; if, later in life, anemia compromises vascular perfusion of vital organs; or if leg ulcers or extramedullary hematopoietic tumors develop. Whether asymptomatic patients with moderate HS should have a splenectomy remains controversial. Partial splenectomy may also be indicated in some of these patients. Splenectomy can be deferred, probably indefinitely, in patients with mild HS and compensated hemolysis. In young symptomatic children with HS, splenectomy should be delayed until at least 3 years of age, and, if possible, until age 6 to 9 years of age, due to the risk of infection. In addition, after 6 years of age, the number of febrile infections is reduced, which decreases the frequency of postsplenectomy fever evaluations.
Surgical Procedures
Laparoscopic Splenectomy.
When splenectomy is warranted, laparoscopic splenectomy is the method of choice in most centers, even for large spleens. The procedure can be combined with laparoscopic cholecystectomy if desired. Comparisons of the laparoscopic approach to the open surgical approach demonstrate fewer complications, less postoperative discomfort, a quicker return to preoperative diet and activities, shorter hospitalization stays, decreased costs, and smaller scars in the laparoscopic group. The drawbacks of a laparoscopic approach are longer operative time, potentially greater difficulty in controlling bleeding, and capsular fracture with subsequent splenosis. Recent innovations include doing the entire laparoscopic procedure through a single incision, which decreases pain and improves the cosmetic result. Nearly all recent studies support the laparoscopic approach to splenectomy as the method of choice in centers in which the surgical staff is experienced.
Partial (Subtotal) Splenectomy.
The emergence of antibiotic-resistant pneumococci and increasing evidence suggesting an increased risk of thrombosis after splenectomy has led to reexamination of the role of alternate treatment modalities. Partial splenectomy has been suggested for selected patients with HS and other hematologic conditions. The goal of the operation is to decrease hemolysis while maintaining residual splenic phagocytic function. In practice, at least 80% to 90% of the enlarged organ is removed ( Fig. 16-21 ). Partial splenectomy was initially an open operation, but laparoscopic and robotic approaches have been developed and are becoming more common. The procedure is more time consuming than a total splenectomy and the time for recovery is longer (4 to 7 days of hospitalization), but studies have shown that partial splenectomy is safe and reduces the rate of hemolysis.
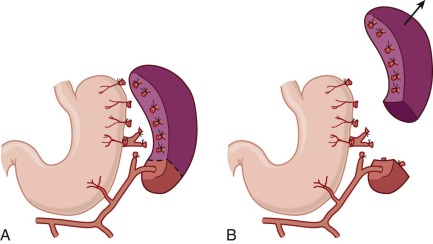
Tchernia and colleagues followed 40 patients for up to 12 years after partial splenectomy and found that the mean hemoglobin value increased from 9.2 g/dL to 12.7 g/dL and the absolute reticulocyte count decreased from 523 × 10 9 cells/L to 267 × 10 9 cells/L ( Fig. 16-22 ). In the children with at least 10 years of follow-up, no differences in hemoglobin and reticulocyte levels were noticed between the group with partial splenectomy and the group with total splenectomy. Significant splenic regrowth was seen in the first year after partial splenectomy, but, after an initial period, the rate of splenic growth, in this study, was much reduced (see Fig. 16-22, C ). Nevertheless, 8% of patients in this study required a total splenectomy after the partial splenectomy, particularly those with severe HS, because of splenic regrowth and resumption of hemolysis. Moreover, 4 of 18 patients in whom the gallbladder was not removed with the initial surgery, required a subsequent cholecystectomy. In a more recent multiinstitutional study of 62 children with a partial splenectomy for HS, only 5% had recurrent hemolysis requiring reoperation and complete splenectomy. Although these results are encouraging, the percentage of HS patients who will require reoperation many years after a partial splenectomy will not be known for several more decades. This is the major risk of the procedure.
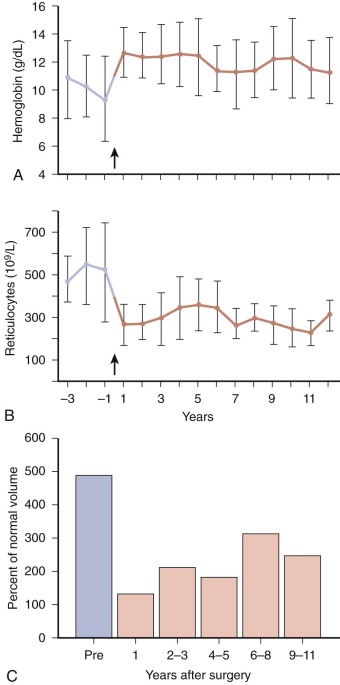
A major argument for partial splenectomy is the expectation that patients will be less susceptible to severe sepsis or thromboembolic complications if some splenic tissue remains. This seems logical and is probably true but is impossible to prove without a very large study over a very long time. We are not aware of any reports of overwhelming sepsis following partial splenectomy, and there is some evidence that the risk of sepsis is less. Experimentally, animals are better protected from bacterial challenge if some splenic tissue remains, though not as well protected as normal animals, and animals and humans recover immune function more rapidly after partial instead of complete splenectomy.
In summary, we believe partial splenectomy is an effective procedure in patients with moderate HS, largely ameliorating hemolysis and anemia while preserving some splenic function. It is important for patients and families to realize that long-term follow-up is encouraging but limited and that splenic regrowth may eventually require a reoperation. For this reason, the procedure cannot yet be recommended as the preferred approach. Some believe that partial splenectomy is the best option for infants with transfusion-dependent HS, who would otherwise be transfused for 3 to 4 years before a complete splenectomy would be done. In such children it is likely that a complete splenectomy will still be needed, however.
Embolization.
A few case reports have reported partial splenic arterial embolization as an alternative to splenectomy to treat hemolysis in patients with HS. Because the experience with this procedure is limited, it cannot be recommended as routine therapy.
Postsplenectomy Changes
After splenectomy, spherocytosis persists but conditioned microspherocytes disappear, and changes typical of the postsplenectomy state, including Howell-Jolly bodies, target cells, siderocytes, and acanthocytes, become evident in the peripheral blood smear. On average, the MCV and mean red cell surface area increase and the MCHC and osmotic fragility decrease, but the effects are modest (5% to 10%). In typical dominant HS, reticulocyte counts fall to normal or nearly normal levels, although the red cell life span, if carefully measured, remains slightly shortened (96 ± 13 days). In all but the most severe occurrences, anemia and jaundice remit and do not recur.
Splenectomy Failure
The rare splenectomy failure (i.e., recurrence of hemolysis) is usually caused by an accessory spleen that was missed during surgery or by another red cell disorder, such as pyruvate kinase deficiency. Accessory spleens occur in 15% to 40% of patients and must always be sought. Occasionally they can be in unusual places: in the chest, across the midline, deeper in the abdomen, and so forth. Recurrence of hemolytic anemia years or even decades after splenectomy should raise suspicion of an accessory spleen, particularly if Howell-Jolly bodies are no longer evident on peripheral blood smears. The absence of “pitted” red cells with craterlike surface indentations, readily seen by interference contrast microscopy, is also a sensitive measure of recrudescent splenic function. The ectopic splenic tissue can be confirmed by a radiocolloid liver/spleen scan or a scan using Cr-labeled, heat-damaged red cells.
Other Therapies
Corticosteroid Therapy
It has been known since the 1950s that large doses of corticosteroids markedly ameliorate HS in unsplenectomized patients, similar to the effects produced by splenectomy. More recent studies have confirmed this finding. Corticosteroids (prednisolone 1 to 2 mg/kg/day or equivalent for 7 days) can be a useful, temporary alternative to splenectomy in controlling severe hemolytic crises.
Vaccination
All candidates for splenectomy should receive a complete series of vaccinations against encapsulated organisms, including pneumococcus, meningococcus, and Haemophilus influenzae . Yearly influenza immunizations are recommended to reduce the chance of secondary bacterial infections. Recent studies have shown that the majority of adults who were splenectomized as children are not up-to-date with their vaccinations. In a study examining the current cases of overwhelming postsplenectomy infection in the era of pneumococcal vaccines, only 31% of 77 patients with such infection received the pneumococcal vaccine. It is important to emphasize the importance of immunizations to the affected parents of children with HS.
Experts differ somewhat on how to protect patients from infection after splenectomy. Our approach follows the 2014 CDC guidelines for vaccination of children and adults with high risk conditions. The guidelines change frequently, so the latest version should be consulted. The recommendations of the British Committee for Standards in Haematology have also been published relatively recently.
If possible, all vaccines should be given at least two weeks and preferably four weeks before splenectomy.
Pneumoccal Vaccines.
All children should receive the 13-valent pneumococcal conjugate vaccine (PCV13, Prevnar 13) beginning at two months in the recommended four-dose schedule. Between two and six years of age, one or two additional doses of PCV13 should be given to children who will have a splenectomy, depending on how many doses of PCV were given in infancy. The minimum interval between doses of PCV13 is 8 weeks. After 2 years of age, the 23-valent pneumococcal polysaccharide vaccine (PPSV23, Pneumovax 23) should also be administered, assuming it hasn’t been given previously. PPSV23 immunization should be delayed until at least 8 weeks after PCV13.
For adults and children 6 years and older who have not received both PCV13 and PPSV23, one dose of the missing vaccine(s) should be given. If both are needed, PCV13 should be given first and followed at least 8 weeks later by PPSV23. If PPSV23 is given first, PCV13 administration must be delayed for at least one year.
A single dose of PPSV23 should be given five years after the first dose and again at 65 years of age.
Haemophilus influenzae Type B Vaccine.
Most children will have received the routine Hib vaccine series and a booster dose prior to the age where splenectomy is considered. Whether fully immunized or not, all patients 15 months of age or older should receive a single dose of a Hib-containing vaccine at least two weeks prior to an elective splenectomy. No studies have been performed on the need for reimmunization, which is not recommended at this time.
Meningococcal Vaccines.
All patients over the age of nine months who are undergoing splenectomy should be immunized with one of the meningococcal conjugate vaccines: MenACWY-CRM (Menveo) or MenACWY-D (Menactra). Both vaccines are targeted at serogroups A, C, W-135, and Y. Serogroups B and X are not covered. Children younger than 19 months of age should receive the four-dose infant series of Menveo. Those 19 through 23 months who have not completed this series should receive two primary doses of Menveo at least three months apart. Adults and children older than two years of age should receive two doses of an MenACWY vaccine at least two months apart. Menactra should not be given until at least four weeks after the completion of all PCV13 doses. Asplenic children whose primary vaccination occurred before 7 years of age should receive an additional dose of either Menveo or Menactra three years later and then every five years. Asplenic adults and children whose primary vaccination occurred after 7 years of age should be revaccinated with Menveo or Menactra every five years.
Antibiotic Prophylaxis
Prophylactic antibiotics after splenectomy are recommended, with emphasis on protection against pneumococcal sepsis (i.e., penicillin V or equivalent, 125 mg orally twice daily in children younger than 5 years of age, and 250 mg twice daily in older children and adults). For those patients allergic to penicillin, erythromycin is recommended. Antibiotic prophylaxis is recommended at least through childhood, and, for teenagers and adults, for at least the first 5 years after surgery.
Some physicians recommend lifelong antibiotic prophylaxis because it is unclear how the risk of postsplenectomy overwhelming infection changes over time. The emergence of penicillin-resistant pneumococci (5% in 1989 to ≥35% in 1997) has led many to reconsider this recommendation, particularly because prior antibiotic use is a risk factor for the development of penicillin resistance in pneumococci. Patients who do not take prophylactic antibiotics should have a supply of oral antibiotics on hand, which they should take immediately should fever higher than 101.5°C develop. They should then seek medical attention immediately.
The risk of infection is decreased in immunized patients, because 50% to 70% of postsplenectomy sepsis is due to S. pneumoniae and about 80% of pneumococcal disease is due to strains contained in the two vaccines. However, it is likely that strains omitted from the vaccines will rise in frequency as pathogens due to selection. Further risk reduction would be anticipated from the chronic use of prophylactic penicillin. Nevertheless, the risk cannot be reduced to zero. Postsplenectomy infections occur occasionally in successfully immunized patients, and compliance with prophylactic medication regimens is a problem, particularly in teenagers.
The occurrence of penicillin-resistant pneumococci is increasing very rapidly all over the world. In some countries, more than one half of all isolates are resistant. In the United States, the prevalence is approximately 35%, but it is much higher in some localities and is rising. Although most of the strains show some sensitivity to penicillin, some are highly resistant and others are multiply resistant. The emergence of these strains will greatly complicate the use of antibiotics for pneumococcal prophylaxis during the next decades.
Folic Acid
Patients with moderate or severe HS should take folic acid (0.5 to 1 mg/day or 5 mg/week orally) prior to splenectomy to prevent folate deficiency. Folate supplementation is unnecessary in mild HS or in HS following splenectomy except during pregnancy.
Animal and Fish Models of Hereditary Spherocytosis and Elliptocytosis
Animal models, such as the well-characterized mouse and zebrafish models, have assisted in our understanding of the pathophysiology of HS. Four types of spontaneous spherocytic hemolytic anemia have been identified in the common house mouse, Mus Musculus : ja/ja (jaundice); sph/sph (spherocytosis) and its alleles (sph 1J /sph 1J [hemolytic anemia], sph 2J /sph 2J [now lost], sph 3J /sph 3J , sph 4J /sph 4J , sph IhJ /sph IhJ , sph 2BC /sph 2BC , and sph DEM /sph DEM ); nb/nb (normoblastosis); and wan/wan. The nomenclature indicates that anemia is observed only in the homozygous state and that the mutants represent four loci: ja, sph, nb, and wan. All the defects are autosomal recessive and cause severe hemolysis, with spherocytosis, jaundice, bilirubin, gallstones, and hepatosplenomegaly, except in the sph DEM /sph DEM mice, which have hereditary elliptocytosis. Another recessive spherocytic hemolytic anemia has been described in the deer mouse, Peromyscus maniculatus . This disorder is less severe and closely resembles the autosomal dominant form of human HS. The spectrin content of deer mouse erythrocytes is reduced by about 20% (unpublished data).
Spectrin Mutants
The ja/ja mutant has a defect in the spectrin β-chain (Arg1160→Stop) and produces no detectable spectrin. The sph/sph variants lack α-chains but have small amounts of β-spectrin. These mice do not produce α-spectrin either due to decreased synthesis, function, or stability. The sph and sph 2BC alleles are frameshift mutations and null alleles. Four mutations affect the C-terminus of α-spectrin, which was previously thought to be functionless in erythrocytes: (1) sph IhJ is a null mutation (Q1853Stop) in repeat 18 ; (2) sph 3J contains a missense mutation (H2012Y) in repeat 19, near the C-terminus, that alters splicing and results in premature termination of translation ; (3) sph 1J allele lacks the last 13 amino acids of α-spectrin ; and (4) sph 4J is a missense mutation (C2384Y). The latter two mutations are in the C-terminal EF-hand domain. All four mutations show marked spherocytosis, and sph 4J does so despite minimally decreased membrane spectrin and normal band 3 levels. Thus the amino acids in the C-terminus of the spectrin tail are functionally important and, it turns out, are critical in attaching spectrin to actin. The sph DEM /sph DEM mouse is missing exon 11 and 46 amino acids near the amino terminus of α-spectrin repeat 5. These mice have elliptocytes, spherocytes, and poikilocytes, and, therefore clinically appear to be a cross between HS and hereditary elliptocytosis (see later).
Cardiac thrombi, fibrotic lesions, and renal hemochromatosis are found in ja/ja and sph/sph mice in adulthood. Transplantation of the hematopoietic cells from sph/sph mice is sufficient to induce thrombotic events in the transplant recipients. In mice with α-spectrin mutations, 70% to 100% experience cerebral infarction. Studies of red cell adhesion in these mice demonstrate a tenfold higher adhesion to thrombospondin compared with normal red blood cells and an increased adhesion to laminin. Therefore it is hypothesized that changes in the red cell adhesive characteristics contribute to the thrombotic events in these mice.
Riesling ( ris ) is an induced mutation in zebrafish that results in a nearly bloodless phenotype, which causes severe anemia and congestive heart failure. The ris phenotype is caused by a null mutation in β-spectrin. The red blood cells of affected zebrafish are spherocytic rather than elliptocytic, the normal zebrafish red cell shape. Scanning electron micrographs show membrane loss, as in other forms of severe HS.
Ankyrin Mutants
Mice with the nb/nb mutation have a primary defect in ankyrin. This is due to a single-nucleotide deletion leading to a frameshift and premature chain termination in the regulatory region of ankyrin. The nb/nb red cells have 50% to 70% of normal spectrin levels but only a trace of ankyrin mRNA and protein. This is in striking contrast to ankyrin defects in human HS, in which ankyrin and spectrin levels are comparably depressed. Fetal nb/nb mice have no anemia and normal reticulocyte counts at birth, possibly because of expression of ankyrin-related proteins in utero. Mice that completely lack Ank1 mostly die in utero. Those that survive have extreme hemolysis and are severely deficient in β-spectrin as well as ankyrin. However, recently a mouse mutation (hema6) has been identified that results in decreased levels of full-length normal ankyrin and more typical HS. This should be useful experimentally.
The nb/nb mice also lack ankyrin in cerebellar Purkinje cells. This leads to an age-related loss of Purkinje cells during the first 5 to 7 months of life and the emergence of cerebellar ataxia. Spinocerebellar degeneration and related syndromes have also been reported in a few adult humans with HS, although it is not yet known whether ankyrin is affected.
Band 3 Mutants
Complete deficiency of band 3 was first described in cattle with a recessive form of HS. In cows, homozygosity for a nonsense mutation in the band 3 gene leads to absence of band 3 and protein 4.2. Red cell membranes are unstable and show surface area loss, as demonstrated by invagination, vesiculation, and extrusion of microvesicles. The affected cattle have defective anion transport and a mild acidosis but mild hemolysis in comparison with other band 3–deficient species.
In mice, targeted disruption of the band 3 gene causes a severe spherocytic hemolytic anemia, with exuberant loss of the membrane surface as vesicles, tubules, and myelin forms. Red cell membranes also lack protein 4.2 and glycophorin A. Surprisingly, the content of membrane skeletal proteins and the architecture of negative-stained, spread skeletons is normal or nearly normal. Therefore band 3 appears to be required for stabilizing membrane lipids but not for membrane skeleton assembly. For unclear reasons, these band 3–deficient mice have a significant propensity for thrombosis and often die in the neonatal period.
Another mouse model, wan/wan , in a C3H/HeJ background, has a null defect in the band 3 gene. These mice have a severe anemia, which is lethal in the neonatal period. However, when wan/wan mice are crossed with wild-type mice of a different strain, Mus castaneus , the F2 generation shows variable hematologic severity. The genetic modifier causing this variability in hematologic abnormalities has been localized to a small chromosomal region containing the β-spectrin gene.
Finally, zebrafish with band 3 mutations, associated with the retsina ( ret ) phenotype, suffer from severe anemia with a complete arrest in erythroid maturation at the late erythroblast stage. These rare ret/ret red blood cells are spherocytic rather than elliptical. Many of these arrested erythroblasts have bilobed nuclei that demonstrate defects in cytokinesis.
Protein 4.2
Mice with targeted deletions of the protein 4.2 gene survive normally and have only mildly spherocytic anemia and hemolysis.
Protein 4.1R
Mice lacking the protein 4.1R gene due to targeted deletion have moderate hemolysis and decreased red cell membrane stability. In addition to lacking protein 4.1R, these mice are also deficient in protein 55, glycoprotein C, ankyrin, and spectrin. The red cell morphology is remarkable for spherocytic shape, rather than elliptocytic shape, likely related to the spectrin deficiency. Interestingly, the knockout mice exhibit specific neurobehavioral deficits in movement, coordination, balance, and learning that correspond with the selective localization of 4.1R in granule cells of the cerebellum and dentate gyrus. Two zebrafish models with protein 4.1R deficiency have been discovered, merlot and chablis, and these mutants have very severe hemolytic anemia, characterized by spherocytosis and increased osmotic fragility.
Adducin and Dematin
Mice lacking just β-adducin or β-adducin and γ-adducin have a very mild, spherocytic hemolytic anemia. Knockout of α-adducin, which eliminates all three adducins (α, β, and γ), still results in only mild hemolysis and moderate spherocytosis, though slightly more severe than in the β-adducin knockout. Similarly, loss of the headpiece of dematin produces, at most, very mild hemolysis. However, mice that lack both β-adducin and the dematin headpiece have a severe spherocytic hemolytic anemia that resembles severe HS. The data suggest the various vertical membrane connections involving the actin junctions—4.1R-p55–glycophorin C, 4.1R–band 3, alpha spectrin–4.2–band 3, adducin–band 3, adducin–Glut1, and actin-dematin–Glut1—are largely redundant and only become consequential when more than one is eliminated.
Hereditary Elliptocytosis and Hereditary Pyropoikilocytosis
Hereditary elliptocytosis (HE) is characterized by the presence of elliptical erythrocytes on peripheral blood smears ( Fig. 16-23 ) and defects in the “horizontal” connections that tie the membrane skeleton together (see Fig. 16-4 ), particularly mutations that impair spectrin self-association. Clinically, these disorders range from the asymptomatic carrier state to severe hemolysis to death in utero due to hydrops fetalis.
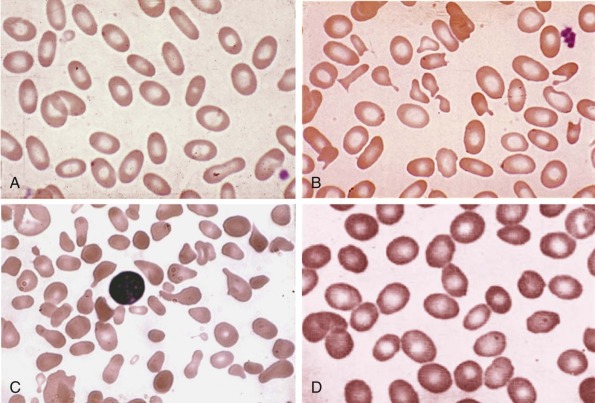
History
HE was first reported in 1904 by Dresbach, a physiologist at Ohio State University, who discovered the condition in one of his histology students during a laboratory exercise in which the students were examining their own blood. This report elicited some controversy, because the student died soon thereafter, leading to speculation that the student actually suffered from incipient pernicious anemia. A number of famous pathologists supported Dresbach’s view that the elliptocytosis was a primary disorder, and this view was substantiated during the next two decades by the reports of Bishop and Huck and Bigelow. The demonstration of the disease in three generations of one family clearly established its hereditary nature.
In the 1930s and 1940s, there was some debate about whether HE was a disease or simply a morphologic curiosity. Early on, some confusion also existed in the differentiation of HE from sickle cell anemia, from hypochromic elliptocytosis (probably thalassemia), and, later, from HS. These reports emphasize the morphologically deceptive nature of HE and its hemolytic variants. Additional historical features of the disease are found in the reports of Wyandt and associates, Wolman and Ozge, and Dacie.
Prevalence and Genetics
In the United States, HE has been estimated to occur in 1 in 2000 to 4000 of the population. The true incidence of HE is unknown because the clinical severity is heterogeneous and many patients are asymptomatic. HE is much more common in areas of endemic malaria, specifically in people of African and Mediterranean ancestry. The higher frequency of HE in these populations suggests that this disease may confer some resistance to malaria. In a study of HE in Benin, Western Africa, an incidence of 1.6% was observed.
HE variants are inherited predominantly in an autosomal dominant manner. Typically, individuals who are heterozygous for an elliptocytic variant have asymptomatic elliptocytosis without anemia. Individuals who are homozygotes or compound heterozygotes for HE variants may suffer from mild to severe hemolysis with moderate to marked anemia. Spontaneous elliptocytogenic mutations have been reported. Presumably, they are also inherited in an autosomal dominant fashion. In one unique kindred with Alport syndrome, mental retardation, midface hypoplasia, and elliptocytosis, inheritance was X-linked and associated with a submicroscopic chromosome X deletion.
HE is heterogeneous with multiple genetic loci, including reports of mutations in α-spectrin, β-spectrin, protein 4.1R, and glycophorin C genes. Missense mutations in α-spectrin or β-spectrin that produce a functional defect in spectrin self-association are the most common. The mutations described include deletions, point mutations, insertions, and splicing defects. The incidence of one common elliptocytic mutation of α-spectrin, α I/65-68 , approaches 1% in Central Africa. It has a worldwide distribution in people of African ancestry. Genetic haplotyping studies suggest that this mutation may have a “founder effect,” originating in central Africa similar to the Benin type of hemoglobin. This observation further supports the possibility that there has been genetic selection for HE because it imparts some resistance to malaria.
Clinical Syndromes
Most of the reported patients with HE can be classified into one of four clinical categories: typical heterozygous HE, HE with hemolysis/hereditary pyropoikilocytosis (HPP), spherocytic HE, and Southeast Asian ovalocytosis (SAO) ( Table 16-11 ). With the exception of SAO, which is homogeneous in molecular terms and is considered separately (see later), these classifications denote clinical phenotypes and not specific molecular causes, although correlations between the two exist.
| Clinical Manifestations |
| Typical Heterozygous HE |
|
| Homozygous HE or HPP |
|
| Spherocytic HE |
|
| Southeast Asian Ovalocytosis |
|
| Laboratory Manifestations |
|
Typical Heterozygous Hereditary Elliptocytosis
The clinical characteristics of typical HE trait vary considerably, defining several clinical subtypes. Different clinical patterns may be seen in members of the same family, and the clinical presentation in single individuals may vary over time, so the clinical patterns are probably more useful for illustrating the spectrum of HE than for classifying the disease.
Silent Carrier State.
This condition was identified by analyzing asymptomatic members of kindreds with HE or HPP. The affected persons have red cells with normal morphologic characteristics and no evidence of hemolysis, but detailed investigations show a subtle defect in their membrane skeletons, with decreased red cell thermal stability, decreased mechanical stability of isolated skeletons, an increased fraction of spectrin dimers in 0°C spectrin extracts, abnormal tryptic peptide maps of spectrin, or various combinations of these defects. Some patients also have α-spectrin LELY , a thalassemia-like splicing defect that impairs production of an otherwise normal α-spectrin protein (see later). It is notable that some patients classified as “silent carriers” have the same molecular defect as patients with mild typical HE, emphasizing the variability of clinical expression.
Typical Hereditary Elliptocytosis (Common or Mild Hereditary Elliptocytosis).
This is the most common clinical form of HE. Patients are asymptomatic, and HE is often diagnosed incidentally when individuals are undergoing screening for an unrelated condition. These persons are not anemic and rarely have splenomegaly. Red cell survival may be normal, but more often there is very mild, compensated hemolysis with a slight reticulocytosis and decreased haptoglobin level. In these patients, HE is hardly more than a morphologic curiosity. The peripheral blood smear shows prominent elliptocytosis with little red cell budding or fragmentation and no spherocytosis. A subset of patients with more functionally severe defects, usually those most affecting spectrin self-association (see later), have slightly greater hemolysis and anemia together with some red cell fragmentation. Elliptocytes (by definition) usually exceed 30% of the red cells and, in some instances, approach 100% (see Fig. 16-23, A ). Very elongated elliptocytes are common (>10%). These patients are easily separated from normal individuals, who have less than 2% to 5% elliptocytes. Somewhat higher proportions of elliptocytes are seen in patients with anemias, particularly megaloblastic anemias, hypochromic-microcytic anemias, myelodysplastic syndromes, and myelofibrosis, but even in these individuals elliptocytes do not exceed 35%. Thus the diagnosis of typical HE is rarely difficult.
Typical Hereditary Elliptocytosis with Sporadic Hemolysis.
Patients with typical heterozygous HE may develop uncompensated hemolysis in response to stimuli that cause hyperplasia of the reticuloendothelial system, particularly if the spleen is involved. Examples include viral hepatitis, cirrhosis, infectious mononucleosis, bacterial infections, and malaria. Hemolysis has also been observed with thrombotic thrombocytopenic purpura and with disseminated intravascular coagulation, which suggests that elliptocytes may be especially susceptible to microangiopathic damage. For unknown reasons pregnancy and cobalamin (vitamin B 12 ) deficiency may also transiently aggravate the disease.
Hereditary Elliptocytosis with Infantile Poikilocytosis.
Infants with typical HE sometimes begin life with moderately severe hemolytic anemia, characterized by marked red cell budding, fragmentation, and poikilocytosis (see Fig. 16-23, B ) and by neonatal jaundice, which may require an exchange transfusion. Usually enough elliptocytes are present to suggest the diagnosis, but this is not always so and the disorder is mistaken for sepsis, infantile pyknocytosis, or a microangiopathic or oxidant-induced hemolytic anemia. HE with infantile poikilocytosis can easily be distinguished from the latter conditions if the parents’ blood smears are examined, because one will show typical HE. However, it is more difficult to distinguish HE with infantile poikilocytosis from severe HE or HPP (see later). Many α-spectrin variants have been associated with the infantile poikilocytosis syndrome. The factors that determine susceptibility are unknown. With time, fragmentation and hemolysis decline, and the clinical picture of typical HE emerges. This transition requires 4 months to 2 years. Subsequently, the disease is clinically indistinguishable from typical HE. The prevalence is unknown, but in our experience, it is unusual but not rare.
The fragmenting neonatal elliptocytes are very sensitive to heat, as are hereditary pyropoikilocytes (see later), but unlike with pyropoikilocytes, this sensitivity lessens over time. During the conversion, the poikilocytic red cells are dense (old) and rich in hemoglobin F whereas the smooth elliptocytes are less dense (young) and enriched in hemoglobin A (unpublished observations). This finding suggests that the change in the disease corresponds to the change from fetal to adult erythropoiesis. No variations in the primary α-spectrin defect or its functional effects on spectrin self-association occur during the conversion, so other skeletal interactions must differ in fetal and adult red cells. Mentzer and associates made the interesting suggestion that 2,3-DPG is the critical agent. Because it is not bound by hemoglobin F, the concentration of 2,3-DPG is elevated in fetal red cells. The free anion is known to weaken spectrin-actin–protein 4.1R interactions but the mechanical properties of normal red cells or ghosts are not greatly impaired at physiologic concentrations of 2,3-DPG in vitro. Whether they are weakened in intact elliptocytes , which are more fragile, is untested. If 2,3-DPG does impair spectrin-actin–4.1R binding in situ, the inherited defect in spectrin self-association would certainly be aggravated. However, if this is the explanation it is not clear why only a subset of infants with HE develop infantile poikilocytosis. Perhaps they are the ones with the more functionally severe mutations.
Hereditary Elliptocytosis with Dyserythropoiesis.
In a small number of families with otherwise typical HE, the sporadic occurrence of hemolysis and anemia is at least partially due to the development of dysplastic and ineffective erythropoiesis. All the reported patients with this rare syndrome are from Italy. They have somewhat more rounded red cells than is typical for HE plus the characteristic findings of ineffective erythropoiesis: relatively low reticulocyte counts, indirect hyperbilirubinemia, and high serum iron and ferritin concentrations. Erythrocytes from some patients are macrocytic. The patients’ bone marrows are hyperplastic, with decreased late erythroblasts and dysplastic features (asynchrony of nuclear-cytoplasmic maturation, binuclearity, internuclear bridges, and small numbers of ringed sideroblasts). Anemia and, presumably, erythroid dysplasia usually commence during adolescence or early adult life and advance gradually over years. Splenectomy is not curative for these patients. The available data suggest that dysplasia and elliptocytosis co-segregate because no individuals with dysplasia have been observed who did not also have elliptocytosis. These families must have a unique subtype of HE because none of the typical HE protein defects were observed in one well-studied family.
Typical Hereditary Elliptocytosis with Chronic Hemolysis and Hereditary Pyropoikilocytosis
These two conditions are really variations of the same thing: homozygosity or compound heterozygosity for elliptocytogenic mutations and/or silent alleles that augment elliptocytogenic mutations. In general, the greater the hemolysis the more the evidence of membrane instability on the peripheral blood smear: budding red cells, fragments, and other bizarre poikilocytes.
Combination of Typical Hereditary Elliptocytosis with α-Spectrin LELY .
Roughly 10% of HE patients have a more severe variant. Sometimes this occurs within members of the same kindred. It is often due to coinheritance of a very common α-spectrin polymorphism that leads to decreased α-spectrin expression—the α LELY allele—in trans to a severely dysfunctional α-spectrin HE allele. The α LELY allele has the effect of increasing the proportion of the HE α-spectrin that is available to combine with β-spectrin and thereby increasing the fraction of defective spectrin on the membrane (see later). Patients with α LELY opposite a severe α-spectrin HE mutation (α LELY /α Severe HE ) have marked hemolysis and elliptopoikilocytosis compared with their siblings with wild-type α-spectrin opposite the same HE allele (+/α Severe HE ), whereas patients with α LELY combined with a mild HE defect (α LELY / α Mild HE ) have more elliptocytes than their siblings with +/α Mild HE but no significant hemolysis. Less often hemolysis is caused by heterozygous inheritance of a mutant spectrin that is grossly dysfunctional. This is indicated by dominant transmission of the hemolytic syndrome and by an unusually high fraction of spectrin dimers in 0°C spectrin extracts (see later).
Hereditary Pyropoikilocytosis.
This uncommon and unfortunately named disorder presents in infancy or early childhood as a hemolytic anemia (hemoglobin level of 2 to 11 g/dL) characterized by extreme poikilocytosis with budding red cells, fragments, spherocytes, triangulocytes, and other bizarre-shaped cells, as well as, in some patients, few or no elliptocytes (see Fig. 16-23, C ). The morphologic characteristics of the blood smear somewhat resemble those seen in patients who have severe thermal burns, which explains the name of the disorder. In fact, it is just a severe variant of HE. Most of the patients are individuals of African origin. Patients typically exhibit hyperbilirubinemia in the neonatal period or marked anemia in the first few months of life, but diagnosis may be delayed in milder variants. Red cell fragmentation, erythroblastosis, and splenomegaly are also characteristic. Complications of severe anemia, including growth retardation, frontal bossing, and early gallbladder disease, have been reported. The red cells are very osmotically fragile, particularly after incubation. In the most severely affected patients, there is a significant microcytosis, with very low MCVs (25 to 75 fL), because of the large number of fragmented red cells. Another characteristic feature of these cells is their remarkable thermal sensitivity. Hereditary pyropoikilocytes fragment at 45°C to 46°C (normal 49°C) after short periods of heating (10 to 15 minutes). After splenectomy, hemolysis is markedly decreased, with the hemoglobin level typically ranging from 10 to 14 g/dL with 3% to 10% reticulocytes.
Some HPP patients are homozygous or compound heterozygous for a structural variant of spectrin, located in the region of the spectrin αβ heterodimer self-association. Rare patients with homozygous protein 4.1R deficiency have a similar picture. Other HPP patients are compound heterozygotes for an α-spectrin mutation and a silent allele such as α LELY or an even more severe thalassemia-like defect of spectrin synthesis. As noted earlier, this thalassemia-like defect enhances the expression of the mutant α HE -spectrin in the cells. An α°-spectrin mutation opposite the α HE -spectrin allele would functionally be the same as having homozygous HE.
Unlike typical HE, HPP red cells are often deficient in spectrin. Presumably if the defect in spectrin self-association is severe enough spectrin either falls off the membrane or never properly assembles. The degree of spectrin deficiency correlates with the severity of HPP and is presumably responsible for the large number of spherocytes and relative paucity of elliptocytes in some patients.
Spherocytic Hereditary Elliptocytosis
This dominant disorder is a phenotypic hybrid of typical HE and HS. It has only been reported in Caucasian families of European descent. The prevalence of spherocytic HE is unknown, but judging from the number of published reports, it is relatively rare, probably accounting for no more than 5% of HE cases in patients of European ancestry. Unlike in typical HE, almost all affected patients have some hemolysis. This is usually mild to moderate and is often incompletely compensated. The elliptocytes are fewer and plumper than in typical HE (see Fig. 16-3, D ), and some spherocytes, microspherocytes, and microelliptocytes are often present. Poikilocytes and red cell fragments are uncommon, a finding that distinguishes this disorder from typical HE with hemolysis. Some spiculated cells may be present following splenectomy, similar to what is seen in β-spectrin deficient HS. Red cell morphologic findings may vary, even within the same family. Some family members may have relatively prominent spherocytes and as few as 10% to 20% elliptocytes, whereas in others elliptocytes predominate and spherocytes are rare. This may cause diagnostic confusion initially, particularly if the propositus has few elliptocytes. Family studies almost always reveal some members with obvious elliptocytosis.
Spherocytic elliptocytes are osmotically fragile, particularly after incubation. Gallbladder disease is common, and aplastic crises are a risk. The pathologic course of spherocytic HE mimics that of HS. Splenic sequestration is evident, red cells are conditioned during splenic passage, and hemolysis abates after splenectomy.
The molecular pathology of classic spherocytic HE is not well studied. Patients with C-terminal truncations of α-spectrin (see later) have many of the clinical features of spherocytic HE and probably are a subset of the disorder. Patients with such truncations typically have moderate hemolysis and anemia, punctuated by recurrent, severe hemolytic crises. Blood smears show plump and usually smooth elliptocytes, although in a few instances, poikilocytosis was prominent.
Patients who lack glycophorin C (see later) also have positive osmotic fragility tests and rounded, smooth elliptocytes. They should probably also be classified as having a recessive (and unusually mild) variant of spherocytic HE. Patients who lack protein 4.2 (another recessive condition) sometimes display features of spherocytic HE (ovalocytes, spherocytes, stomatocytes) ; however, protein 4.2 deficiency more often resembles HS, morphologically and pathophysiologically, and is better classified as a variant of that disorder. Mice that lack tropomodulin-1 have mild spherocytic HE that is more spherocytic than elliptocytic. The anemia may be partly compensated by an increase in tropomodulin-3, which is not normally expressed in mature red cells, to about 20% of the normal level of tropomodulin-1. Tropomodulin deficiency has not been identified in humans but should be considered.
Very rare patients appear to be compound heterozygotes for HS and HE. One Turkish girl is a particularly good candidate. Her mother had mild HS and her father most likely had very mild HE, whereas she suffered from a moderately severe hemolytic anemia (hemoglobin level, 8.4 g/dL; reticulocyte count, 24%; bilirubin level, 1.6 mg/dL), with frontal bossing, osteoporosis, splenomegaly, and a mixture of microspherocytes and rounded elliptocytes.
Southeast Asian Ovalocytosis
This condition, which has a unique phenotype, molecular defect, and geographic distribution, is discussed later in this chapter.
Treatment
Typical HE is mild and splenectomy is not required. Serial interval ultrasound examinations, beginning at approximately 6 years of age, to detect gallstones should be performed in patients with brisk hemolysis. Splenectomy to decrease hemolysis, ameliorate anemia, and avoid the formation of bilirubinate gallstones has been the cornerstone of therapy for patients with severe hemolytic HE and HPP. Most practitioners believe that the indications for splenectomy in HS should also be applied to patients with symptomatic HE and HPP. Patients with HE and HPP who have been splenectomized have experienced increased hematocrit values, decreased reticulocytosis, and improvement in clinical symptoms. The risk of thrombosis after splenectomy for HE is unclear, although cases have been reported. If hemolysis is still active after splenectomy, folic acid should be administered daily. Recommendations for antibiotic prophylaxis, immunization, and monitoring during intercurrent illnesses are the same as those noted for HS patients before and after splenectomy.
Neonates with infantile poikilocytosis should be treated as any patient with hemolytic anemia. Phototherapy and exchange transfusions are warranted in neonates with severe anemia and pathologic hyperbilirubinemia. Splenectomy is never indicated in the neonatal period. HE patients with extreme hemolysis should be transfused until they are 2 to 3 years of age, when immunizations are effective and splenectomy can be performed safely. Partial splenectomy is not as effective as in HS.
Laboratory Methods
Thermal Sensitivity of Red Cells and Spectrin
Red cells heated to temperatures approaching 50°C for short periods of time become unstable and fragment spontaneously, probably owing to denaturation of spectrin. Normal spectrin denatures at 49°C (10-minute exposure), and normal red cells fragment at the same temperature. Almost all patients with HPP and some patients with milder forms of HE have thermally sensitive red cells. Red cells from patients with HPP and infants with HE and infantile poikilocytosis fragment after 10 minutes at 44 to 46°C. Red cells from some but not all patients with typical HE fragment at 47 to 48°C. As expected, purified spectrin from these red cells is also heat sensitive. This test is limited because we do not understand, in molecular terms, why specific mutations are thermally sensitive. However, it remains one of the simplest tests available for diagnosing HPP in laboratories that do not specialize in membrane protein analysis.
Abnormal Spectrin Oligomerization
In many patients with HE and all patients with HPP, spectrin dimers are not properly converted to tetramers and higher oligomers in vitro or on the membrane. This important functional property is easily assessed in low-temperature spectrin extracts. At 0°C, the equilibrium between spectrin dimer and tetramer is kinetically frozen. If spectrin is extracted from the membrane at 0°C and carefully protected from warming during separation of dimers, tetramers, and oligomers (usually on nondenaturing polyacrylamide gels), the proportion of each spectrin species reflects its relative proportion on the membrane. Patients with defects in spectrin self-association have abnormally high proportions of spectrin dimer in 0°C spectrin extracts (i.e., more than 10% of total spectrin dimers and tetramers). The fraction of spectrin dimers is an important functional assessment in patients with α-spectrin defects. It correlates well with clinical severity and accurately predicts unusually severe mutations. Conversely, discordance between the degree of hemolysis and fraction of spectrin dimers may alert the physician to an underlying, secondary complication.
Ektacytometry
The technique of osmotic gradient ektacytometry (OGE) to measure membrane surface area and cell hydration of intact red cells was discussed earlier, in the section on laboratory tests in HS, but the ektacytometer can also be used to assess red cell membrane mechanical stability. Isolated red cell ghosts are subjected to high shear stress in a laser diffraction viscometer, and the DI (a measure of the average elongation of the sheared ghosts) is recorded as a function of time. Fragile ghosts fragment into small spheres more quickly than normal, causing their “deformability” to fall. The technique is a useful screening test because membrane stability is reduced in HE and dramatically so in HPP.
Eosin-5′-Maleimide–Binding Test
EMA binds to band 3 and members of the Rh protein complex and is a measure of the amount of these proteins on the red cell surface, and therefore an indirect measure of membrane surface area (see earlier discussion under Hereditary Spherocytosis ). Because red cell surface area is greatly decreased by cell fragmentation in HPP, EMA fluorescence is very reduced on a per cell basis, even more than it is in HS.
Tryptic Maps of Spectrin
Limited tryptic digestion of spectrin extracted from erythrocytes, followed by two-dimensional gel electrophoresis, separates the resulting trypsin-resistant domains of α-spectrin and β-spectrin, including an 80-kD α-I domain peptide (α I/80 ) containing the self-association site of α-spectrin. Many of the α-spectrin HE mutations reside in this domain and alter its structure so that it is cleaved into smaller fragments by trypsin. These were given names such as α I/74 or α I/65-68 . Although the technique is historically important it is rarely used now.
Genetic Analyses
Increasingly HE mutations are identified by DNA sequencing. Unlike HS, some of the HE mutations are quite common and their location and nature relate to their clinical severity, so it is useful to know the specific molecular lesion. It is also important to know whether the patient carries the common spectrin α LELY polymorphism and whether it is in cis or trans to the HE mutation because that can greatly affect the clinical picture (see below). Typically exons are amplified from genomic DNA isolated from peripheral blood leukocytes, but because most HE mutations are missense mutations, reverse-transcribed reticulocyte or bone marrow mRNA can also be amplified. At the moment, DNA testing for HS and HE mutations is not available commercially and only a few research laboratories will do the analysis. However, with the rapid fall in the cost of whole-exome and targeted exome sequencing, it is likely these will become the methods of choice in the near future.
Pathophysiology
Spectrin Defects
Abnormalities of either α-spectrin or β-spectrin are responsible for most cases of HE and HPP. The majority are due to missense mutations near the spectrin self-association site (blue symbols in Figs. 16-6 and 16-8 ). The spectrin self-association contact site is a combined triple helical “spectrin repeat” (see Fig. 15-13 and Fig. 15-14 ) in which two of the three helices that make up a typical spectrin repeat, helices A and B, are contributed by the C-terminus of the β-spectrin, whereas the third helix (C) comes from the first portion of the NH 2 -terminus of α-spectrin. These three helices pair up and create the bond responsible for spectrin self-association. HE mutations weaken the ability of spectrin dimers to form tetramers and destabilize the membrane skeleton. The functional damage is manifested by an increased proportion of spectrin dimers in 0°C low–ionic strength extracts of spectrin, and by decreased conversion of spectrin dimers to tetramers in solution and on the membrane. The broken spectrin-spectrin links are visible in electron micrographs and diminish the resistance of the isolated membranes to shear stress. The clinical severity of HE correlates with the degree of impairment of spectrin self-association and with the amount of mutant spectrin that is produced.
A few examples of mutations that cause HE or HPP follow. Readers who are interested in a more extensive list of HE/HPP mutations can refer to an earlier chapter by the author or to selected reviews, or consult the paid or free versions of the Human Gene Mutation Database.
Alpha-Spectrin Defects Within the Self-Association Binding Site.
α-Spectrin mutations account for 48% of the reported defects in HE and HPP. Many of the mutations are located in the C helix at the beginning of the α-spectrin chain ( Fig. 16-24 and see Fig. 16-6 ), which is a partial spectrin repeat, also called spectrin repeat 0 (see Fig. 16-24 ). Most of these mutations change amino acids that are part of the contact surfaces between the α-spectrin and β-spectrin chains at the tetramerization site and greatly impair or even abolish spectrin-spectrin binding. As a result, they are among the most severe of the common α-spectrin mutations that cause HE and HPP.