Introduction
Maintenance of the tonicity of extracellular fluids within a very narrow range is crucial for proper cell function. Extracellular osmolality regulates cell shape, as well as intracellular concentrations of ions and other osmolytes. Furthermore, proper extracellular ionic concentrations are necessary for the correct function of ion channels, action potentials, and other modes of intercellular communication. Extracellular fluid tonicity is regulated almost exclusively by the amount of water intake and excretion, whereas extracellular volume is regulated by the level of sodium chloride intake and excretion. In children and adults, normal blood tonicity is maintained over a 10-fold variation in water intake by a coordinated interaction among thirst, vasopressin, and renal systems. Dysfunction in any of these systems can result in abnormal regulation of blood osmolality, which if not properly recognized and treated may cause life-threatening dysfunction in neuronal and other cellular activities.
The posterior pituitary, or the neurohypophysis, secretes the nonapeptide hormones vasopressin (also termed antidiuretic hormone [ADH] ) and oxytocin. Vasopressin controls water homeostasis, and oxytocin regulates smooth muscle contraction during parturition and lactation. Disorders of vasopressin secretion and action lead to clinically important derangements in water metabolism. In this chapter, the physiology of water and volume regulation is summarized, a symptom-based approach to the differential diagnosis of the diseases of water homeostasis is presented, and a review of the pathology and treatment of disorders involving these systems is provided.
Physiology of osmotic and volume regulation
The control of plasma tonicity and intravascular volume involves a complex integration of endocrine, neural, and paracrine pathways. Osmotic sensor and effector pathways control the regulation of vasopressin release and signal transduction, whereas volume homeostasis is determined largely through the action of the renin-angiotensin-aldosterone system, with contributions from both vasopressin and the natriuretic peptide family. An improved understanding of the anatomic structures and molecules involved has developed through recent molecular biologic and physiologic studies.
Osmotic Sensor and Effector Pathways
Vasopressin and Oxytocin Biochemistry
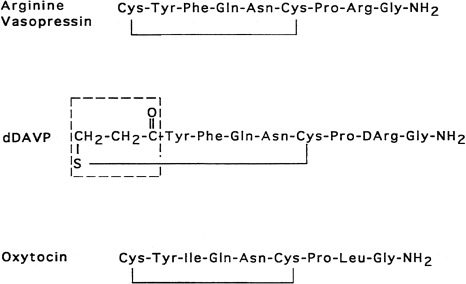
Vasopressin and oxytocin are evolutionarily-related peptides (paralogs), having arisen from gene duplication of a phylogenetically common molecule approximately 450 million years ago. Both peptides consist of a 6-amino-acid disulfide ring, plus a 3-amino-acid tail, with amidation of the carboxy terminus. As early as 1895, a potent biologic principle—consisting of vascular pressor activity, “birth quickening,” and milk secretory effects—was recognized in neurohypophyseal extracts. The sequences of the individual peptides with pressor and antidiuretic capacity (vasopressin) and oxytocic capacity were determined by du Vigneaud and colleagues during the mid-1950s, culminating in the synthesis of each hormone in its biologically active form. In most mammals, vasopressin and oxytocin differ in only two amino acids—one substitution within the ring and one within the tail structure ( Fig. 12.1 ). Exploration of the structure-function relationship of specific amino acids, within both vasopressin and oxytocin, has allowed characterization of molecules with important clinical use. Although the vasopressor activity was markedly diminished by the replacement of l-arginine with d-arginine at position 8 of the vasopressin molecule, the duration of action was prolonged, and the antidiuretic action enhanced by the amino-terminal deamidation, creating the analogue desmopressin (desamino-d-arginine vasopressin [dDAVP]) (see Fig. 12.1 ). Whereas the antidiuretic:pressor ratio for vasopressin is 1:1, this ratio for dDAVP is between 2000:1 and 3000:1. This difference is almost entirely caused by the differential binding of dDAVP to the V2 and V1 receptors (2054:1). dDAVP, with an antidiuretic potency between 2 times and 3 times that of its parent vasopressin, is now routinely used in clinical practice.
The association of vasopressin and oxytocin with specific proteins, the neurophysins, while stored in the neurohypophysis was apparent as early as 1900. Subsequent isolation and characterization of the neurophysins revealed two distinct forms, one type exclusively associated with vasopressin and the other exclusively associated with oxytocin. Both are single-polypeptide chains of molecular weight 10,000 daltons. Despite extensive biophysical characterization, including crystallography of the oxytocin-neurophysin complex, the biologic function of the neurophysins remains unknown. Possible roles for the neurophysins include hormone stabilization against degradation during intracellular storage, more efficient packaging within secretory granules, enhancement of posttranslation processing by the proenzyme convertases, and hormone transport in blood. Recently, copeptin, a segment of vasopressin-associated neurophysin, has been developed as an indicator of vasopressin secretion.
The common origin of vasopressin and its neurophysin from a single larger precursor was first proposed by Sachs and colleagues, who showed increased incorporation of 35S cysteine, infused into canine third ventricle, into vasopressin isolated from the hypothalamus compared with vasopressin isolated from the posterior pituitary. Isolation of the larger precursor from the hypothalamus, followed by trypsin digestion, produced fragments of size similar to that of vasopressin and its neurophysin, with vasopressin immunoreactivity in the 1000-dalton component.
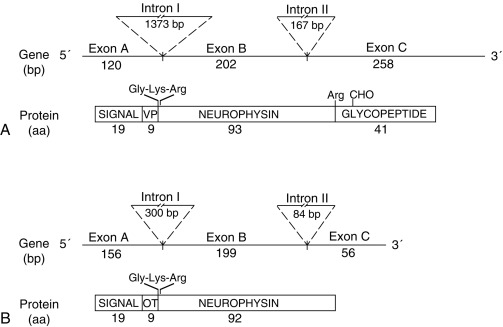
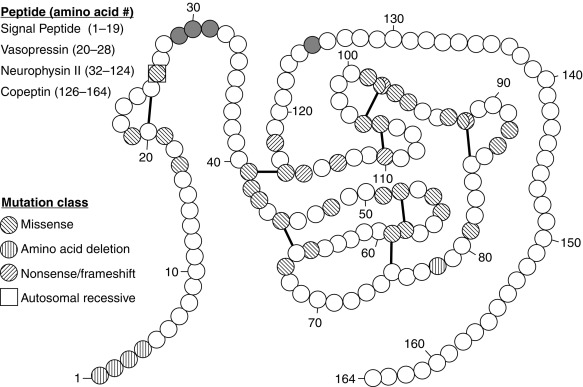
Since 1990, molecular genetic analyses have enhanced the understanding of the synthesis, the processing, and the evolution of the vasopressin and oxytocin preprohormones. All mammalian vasopressin and oxytocin genes each consist of three exons ( Fig. 12.2 ). The first exon encodes the 19-amino-acid signal peptide, followed by vasopressin or oxytocin nonapeptides. This is followed by a 3-amino-acid protease cleavage site, leading into the first nine amino acids of neurophysin II (for vasopressin) or neurophysin I (for oxytocin). After interruption of the coding region by an intron, exon 2 continues with neurophysin coding sequences. The third exon completes the sequence of the neurophysin and, for vasopressin only, is followed by coding information for an additional 39-amino-acid glycopeptide (copeptin) whose function is unclear. Preprovasopressin contains 16 cysteines, which likely participate in eight disulfide bridges that determine the tertiary structure of the protein ( Fig. 12.3 ). One cysteine pair is present in vasopressin peptide, whereas the rest are in neurophysin.
In all mammalian species analyzed thus far, oxytocin and vasopressin genes are adjacent in chromosomal location (chromosome 20 in the human 19) and linked tail to tail, in opposite transcriptional orientation. In the human, they are separated by only 12 kb. This likely explains their origin from the ancient duplication of a common ancestral gene. Whether this adjacent linkage is of regulatory significance is unclear.
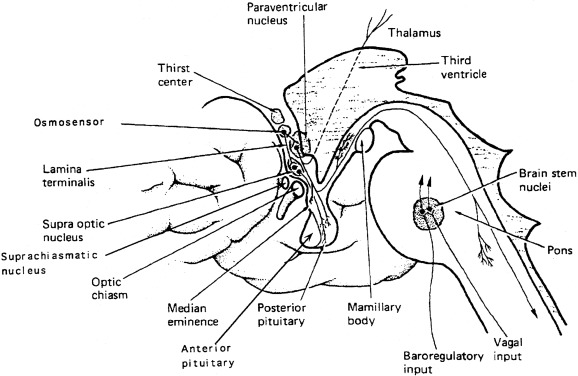
Expression of vasopressin and oxytocin genes occurs in the hypothalamic paraventricular and supraoptic nuclei. The magnocellular components of each of these nuclei are the primary neuronal populations involved in water balance, with vasopressin synthesized in these areas carried by means of axonal transport to the posterior pituitary, its primary site of storage and release into the systemic circulation ( Fig. 12.4 ). The bilaterally paired hypothalamic paraventricular and supraoptic nuclei are separated from one another by relatively large distances (approximately 1 cm). Their axons course caudally, converge at the infundibulum, and terminate at different levels within the pituitary stalk and the posterior pituitary gland (see Fig. 12.4 ). Vasopressin is also synthesized in distinct parvocellular neurons of the paraventricular nucleus, where it has a role in modulation of hypothalamic-pituitary-adrenal axis activity. In this site, vasopressin is colocalized in cells that synthesize corticotropin-releasing hormone, and both are secreted at the median eminence and carried through the hypothalamic-hypophyseal portal capillary system to the anterior pituitary, where together they act as the major regulators of adrenocorticotropic hormone synthesis and release. Vasopressin is also present in the hypothalamic suprachiasmatic nucleus, the circadian pacemaker of the body, where its function is unknown.
Regulation of Vasopressin Secretion and Thirst
Osmotic Regulation
The rate of secretion of vasopressin from the paraventricular and supraoptic nuclei is influenced by several physiologic variables, including plasma osmolality and intravascular volume, as well as nausea and a number of pharmacologic agents. The major osmotically active constituents of blood are sodium, chloride, and glucose (with insulin deficiency). Normal blood osmolality ranges between 280 and 290 mOsm/kg H 2 O.
The work of Verney first demonstrated the relationship of increased vasopressin release in response to increasing plasma osmolality, as altered by infusion of sodium chloride or sucrose. At that time, it was postulated that there existed intracranial sensors sensitive to changes in plasma osmolality. Multiple researchers have subsequently confirmed that plasma vasopressin concentration increases in response to increasing plasma tonicity, with the location of the osmosensor, likely to be within the circumventricular organ, neuronal nuclei surrounding the third ventricle, which lacks. a blood-brain barrier. The organ vasculosum of the lamina terminalis (OVLT) and the subfornical organ (SFO), areas of the preoptic hypothalamus outside the blood-brain barrier, are likely sites of both osmosensing, because lesions of the OVLT result in impaired vasopressin secretion and hypernatremia. Also the site of action of angiotensin II, infused intracerebrally or peripherally, to produce vasopressin secretion and antidiuresis resides within the OVLT.
The pattern of secretion of vasopressin into blood has been characterized extensively in normal individuals and in those with abnormalities in water homeostasis. Normally, at a serum osmolality of less than 280 mOsm/kg, plasma vasopressin concentration is at or below 1 pg/mL, the lower limit of detection of most radioimmunoassays. Above 283 mOsm/kg—the normal threshold for vasopressin release—plasma vasopressin concentration increases in proportion to plasma osmolality, up to a maximum concentration of about 20 pg/mL at a blood osmolality of approximately 320 mOsm/kg ( Fig. 12.5 ). The osmosensor can detect as little as a 1% change in blood osmolality. Plasma concentrations in excess of 5 pg/mL are also found with nausea, hypotension, hypovolemia, and insulin-induced hypoglycemia, but further increments in urine concentration do not occur, because peak antidiuretic effect is achieved at 5 pg/mL. The rate of increase of plasma vasopressin concentration, and thus the sensitivity of the osmosensor, exhibits substantial (as much as 10-fold) interindividual variation as plasma osmolality increases. The set-point for vasopressin secretion varies in a single individual, in relation to changes in volume status and hormonal environment (e.g., pregnancy ) or glucocorticoid status. After the seventh week of gestation, osmotic thresholds for both vasopressin release and thirst are reduced by approximately 10 mOsm/kg (see Fig. 12.5 ), such that normal blood osmolality, during pregnancy, is approximately 273 mOsm/ kg (serum sodium 135 mEq/L). Similarly, thresholds for vasopressin release and thirst during the luteal phase of the menstrual cycle are approximately 5 mOsm/kg lower than those in the follicular phase. Human chorionic gonadotropin, during pregnancy, and luteinizing hormone, during the second half of the menstrual cycle, may contribute to these changes in osmotic thresholds.
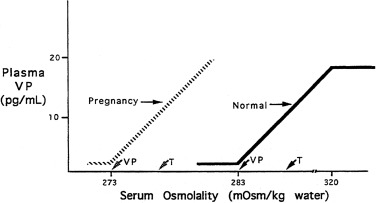
The sensation of thirst, a more integrated cortical activity, is determined by other anatomically distinct hypothalamic neurons, with afferents involving the ventromedial nucleus, and subfornical organ. The activation of the thirst mechanism is probably mediated by angiotensin II. Whether the osmosensor for thirst and vasopressin release are the same is not certain, although this is suggested by lesions in the anteroventral region of the third ventricle that abolish both thirst sensation and vasopressin release. It makes physiologic sense that the threshold for thirst (293 mOsm/kg) is approximately 10 mOsm/kg higher than that for vasopressin release (see Fig. 12.5 ). Otherwise, during the development of hyperosmolality, the initial activation of thirst and water ingestion would result in polyuria, without activation of vasopressin release, causing a persistent diuretic state. Immediately after water ingestion, before a change in blood osmolality or volume, vasopressin concentration falls and thirst ceases. The degree of suppression is directly related to the coldness and volume of the ingested fluid. Water that bypasses the pharynx does not have this effect. The effect is probably mediated by chemoreceptors present in the oropharynx that project to the subfornical organ, which results in decreased thirst and vasopressin release. This “presystemic” regulation of thirst and vasopressin secretion, before osmolality changes, guards against both the rapid overdrinking of fluids after prior intense thirst, and reversal of antidiuresis with the onset of water ingestion.
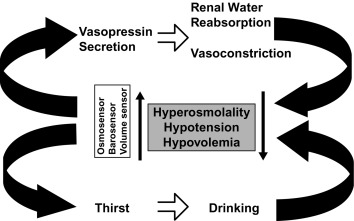
As noted earlier, water balance is regulated in two ways: (1) vasopressin secretion stimulates water reabsorption by the kidney, thereby reducing future water loss, and (2) thirst stimulates water ingestion, thereby restoring previous water loss. Ideally, these two systems work in parallel to efficiently regulate extracellular fluid tonicity ( Fig. 12.6 ); however, each system by itself can maintain plasma osmolality in the near-normal range. For example, in the absence of vasopressin secretion but with free access to water, thirst drives water ingestion up to the 5 to 10 L/m 2 of urine output seen with vasopressin deficiency. Conversely, an intact vasopressin secretory system can compensate for some degree of disordered thirst regulation. When both vasopressin secretion and thirst are compromised, however, by either disease or iatrogenic means, there is great risk of the occurrence of life-threatening abnormalities in plasma osmolality.
Nonosmotic Regulation
Separate from osmotic regulation, vasopressin has been shown to be secreted in response to alterations in intravascular volume. Afferent baroreceptor pathways arising from the right and left atria and the aortic arch (carotid sinus) are stimulated by increasing intravascular volume and stretch of vessel walls, and they send signals through the vagus and glossopharyngeal nerves, respectively, to the brain stem nucleus tractus solitarius. Noradrenergic fibers from the nucleus tractus solitarius synapse on the hypothalamic paraventricular nucleus and the supraoptic nucleus and, on stimulation, inhibit vasopressin secretion.
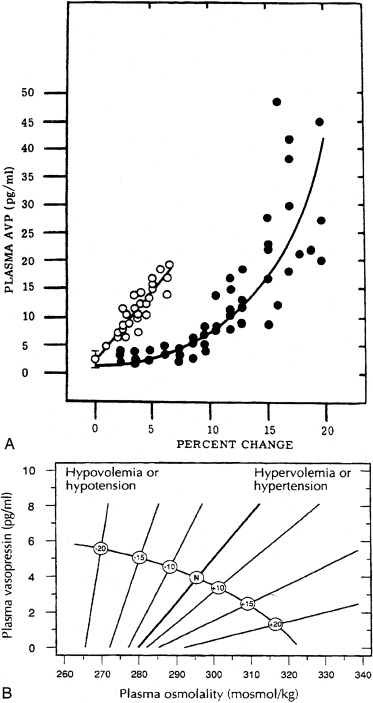
The pattern of vasopressin secretion in response to volume, as opposed to osmotic stimuli, is markedly different ( Fig. 12.7 ). Although minor changes in plasma osmolality above 280 mOsm/kg evoke linear increases in plasma vasopressin, substantial alteration in intravascular volume is required for alteration in vasopressin output. No change in vasopressin secretion is seen until blood volume decreases by approximately 8%. With intravascular volume deficits exceeding 8%, vasopressin concentration increases exponentially. Furthermore, osmotic and hemodynamic stimuli can interact in a mutually synergistic fashion, so that the response to either stimulus may be enhanced by the concomitant presence of the other (see Fig. 12.7 ). When blood volume or blood pressure decreases by approximately 25%, vasopressin concentrations are evident of 20- to 30-fold above normal, and vastly exceeding those required for maximal antidiuresis. Mice with deletion of the V1a vasopressin receptor gene, Avpr1 , indicate that vasopressin acting through this receptor is required to maintain normal blood pressure, even at low concentrations of blood vasopressin.
Nausea, as evoked by apomorphine, motion sickness, or vasovagal reactions, is a very potent stimulus for vasopressin secretion. This effect is likely mediated by afferents from the area postrema of the brain stem and may result in vasopressin concentrations two to three orders of magnitude above basal levels. Nicotine is also a strong stimulus for vasopressin release. These pathways probably do not involve osmotic or hemodynamic sensor systems, because blockade of the emetic stimulus, with dopamine or opioid antagonists, does not alter the vasopressin response to hypernatremia or hypovolemia.
Vasopressin secretion is inhibited by glucocorticoids; because of this, loss of negative regulation of vasopressin secretion occurs in the setting of primary or secondary glucocorticoid insufficiency. The insertion of aquaporin-2 (AQP2) in the renal luminal membrane is stimulated by nitric oxide. Glucocorticoids inhibit nitric oxide synthase, thereby providing a mechanism by which they may facilitate free water clearance, by inhibiting nitric oxide-induced insertion of AQP2 into the luminal membrane. The effects of cortisol loss of both enhancing hypothalamic vasopressin production and directly impairing free water excretion are important considerations in the evaluation of the patient with hyponatremia, as is subsequently discussed.
Vasopressin Metabolism
Once in the circulation, vasopressin has a half-life of only 5 to 10 minutes, owing to its rapid degradation by a cysteine amino-terminal peptidase called vasopressinase . A synthetic analogue of vasopressin, desmopressin, is insensitive to amino-terminal degradation and thus has a much longer half-life of 8 to 24 hours. During pregnancy, the placenta secretes increased amounts of this vasopressinase, resulting in a 4-fold increase in the metabolic clearance rate of vasopressin. Normal women compensate with an increase in vasopressin secretion, but women with preexisting deficits in vasopressin secretion or action, or those with increased concentrations of placental vasopressinase, associated with liver dysfunction or multiple gestations, may develop diabetes insipidus in the last trimester, which resolves in the immediate postpartum period. As expected, this form of diabetes insipidus responds to treatment with desmopressin but not with vasopressin.
Sites of Vasopressin Action
Vasopressin Receptors
Vasopressin, released from the posterior pituitary and the median eminence, affects the function of several tissue types by binding to members of a family of G protein–coupled cell surface receptors, which subsequently transduce ligand binding into alterations of intracellular second messenger pathways. Biochemical and cell biologic studies have defined three receptor types, designated V1, V2, and V3 (or V1b). The major sites of V1 receptor expression are on vascular smooth muscle and hepatocytes, where receptor activation results in vasoconstriction and glycogenolysis, respectively. The latter activity may be augmented by stimulation of glucagon secretion from the pancreas. The V1 receptor on platelets also stimulates platelet aggregation. V1 receptor activation mobilizes intracellular calcium stores through phosphatidylinositol hydrolysis. Despite its initial characterization as a powerful pressor agent, the concentration of vasopressin needed to significantly increase blood pressure is several-fold higher than that required for maximal antidiuresis, although substantial vasoconstriction in renal and splanchnic vasculature can occur at lower concentrations. The cloning of the V1 receptor has greatly elucidated the relationship of the vasopressin (and oxytocin ) receptors and, through sensitive in situ hybridization analysis, has further localized V1 expression to the liver and the vasculature of the renal medulla, as well as to many sites within the brain, including the hippocampus, the amygdala, the hypothalamus, and the brain stem. Compared with their normal counterparts, mice genetically modified to be deficient in the V1 receptor (V1a KO) have been found to have insulin resistance, increased hepatic glucose production, decreased hepatic glycogen content, and decreased aldosterone secretion, despite a lower plasma volume, lower basal blood pressure, greater degree of lipolysis, and impaired nuclear transport of the renal tubular mineralocorticoid receptor. The V3 (or V1b) receptor is present on corticotrophs in the anterior pituitary and acts through the phosphatidylinositol pathway to increase adrenocorticotropic hormone secretion. Its binding profile for vasopressin analogues resembles more closely that of the V1 than the V2 receptor. The structure of this receptor has been determined in humans by cloning of its complementary deoxyribonucleic acid (DNA). Its structure is similar to that of the V1 and oxytocin receptors, and it is expressed in the kidney, as well as in the pituitary. Mice with deletion of the V1b (V3) receptor gene ( V1bKO ) have been created and studied. As expected, they have defective activation of the pituitary-adrenal axis, following some acute and chronic stressors. Male V1bKO mice were also found to have decreased aggression and social motivation.
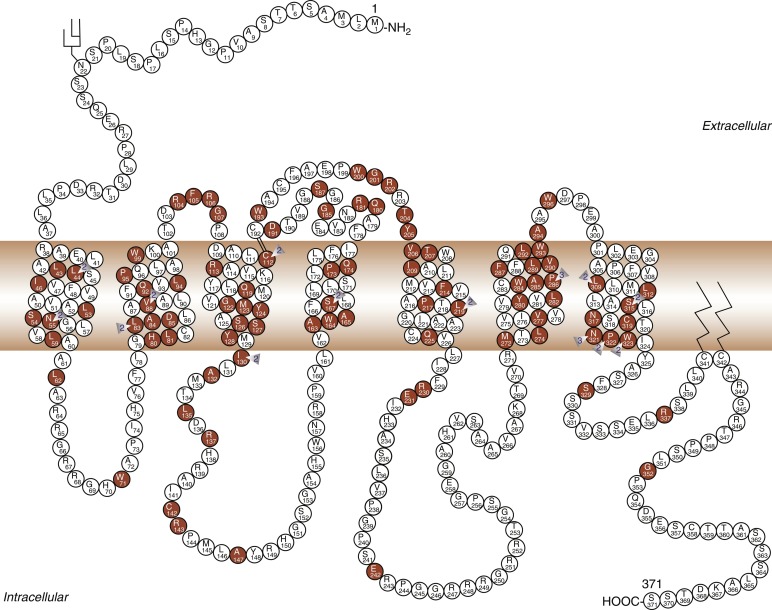
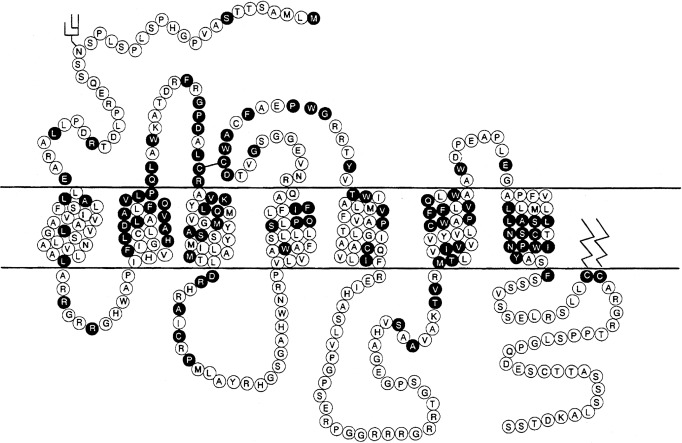
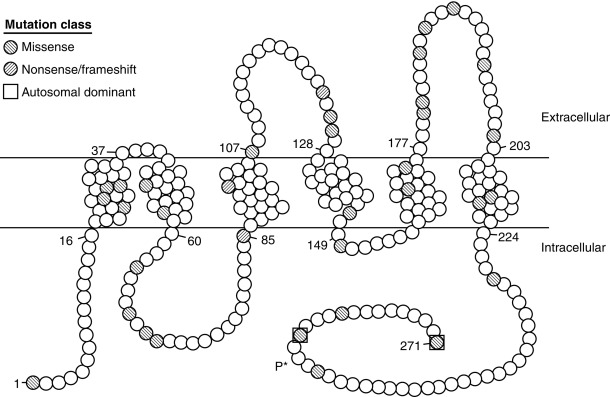
Modulation of water balance occurs through the action of vasopressin on V2 receptors, located primarily in the renal collecting tubule, along with other sites in the kidney, including the thick ascending limb of the loop of Henle and periglomerular tubules. It is also present on vascular endothelial cells in some systemic vascular beds, where vasopressin stimulates vasodilation, possibly through activation of nitric oxide synthase. Vasopressin also stimulates von Willebrand factor, factor VIIIa, and tissue plasminogen activator through V2-mediated actions. Because of this, desmopressin is used to improve the prolonged bleeding times characteristic of uremia, type I von Willebrand disease, and hemophilia. The V2 receptor consists of 370 amino acids encoding seven transmembrane domains characteristic of the G protein–coupled receptors. These transmembrane domains share approximately 60% sequence identity with the V1 receptor, but substantially less with other members of this family ( Fig. 12.8 ). Unlike the V1 and V3 receptors, the V2 receptor acts through adenylate cyclase to increase intracellular cyclic adenosine monophosphate (AMP) concentration. The human V2 receptor gene is located on the long arm of the X chromosome (Xq28), at the locus associated with congenital, X-linked vasopressin-resistant diabetes insipidus. Mice in which V2R has been deleted have a similar nephrogenic diabetes insipidus phenotype.
Renal Cascade of Vasopressin Function
Vasopressin-induced increases in intracellular cyclic AMP, as mediated by the V2 receptor, triggers a complex pathway of events resulting in increased permeability of the collecting duct to water and efficient water transit across an otherwise minimally permeable epithelium ( Fig. 12.9 ). Activation of a cyclic AMP-dependent protein kinase imparts remodeling of cytoskeletal microtubules and microfilaments that culminate in the insertion of aggregates of water channels into the apical membrane. These mechanisms may involve a vesicle-associated membrane protein-2–like protein (VAMP-2), which also regulates synaptic vesicle activity in neuronal terminals, and its associated receptor syntaxin-4.
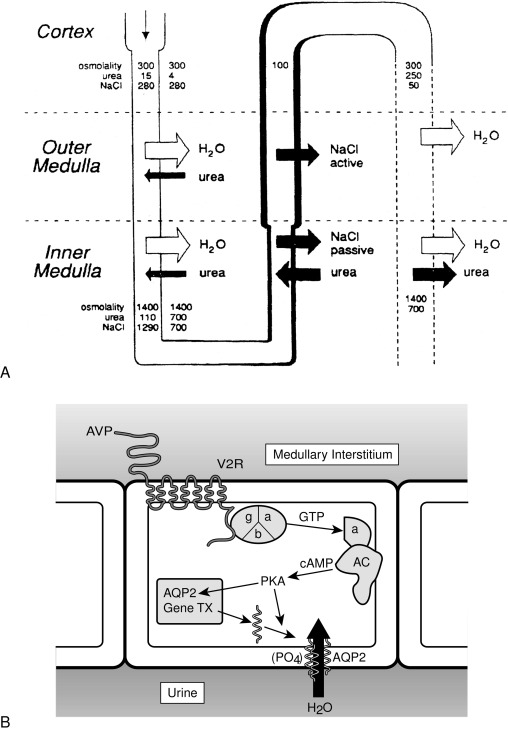
Insertion of the water channels causes an up to 100-fold increase in water permeability of the apical membrane, allowing water movement along its osmotic gradient into the hypertonic inner medullary interstitium from the tubule lumen and excretion of a concentrated urine (see Fig. 12.9 ). The molecular analysis of the water channels has revealed a family of related proteins, designated aquaporins, that differ in their sites of expression and pattern of regulation. Each protein consists of a single polypeptide chain with six membrane-spanning domains ( Fig. 12.10 ). Although functional as monomers, they form homotetramers in the plasma membrane.
AQP2 is expressed mostly within the kidney, primarily within the collecting duct. It is also expressed in the vas deferens, at least in the rat, although it is not regulated by vasopressin in this location. Studies with immunoelectron microscopy have demonstrated large amounts of AQP2 in the apical plasma membrane and subapical vesicles of the collecting duct, consistent with the “membrane shuttling” model of water channel aggregate insertion into the apical membrane after vasopressin stimulation. Studies analyzing the mechanism by which AQP2 traffics to the apical plasma membrane have demonstrated that vasopressin-induced, protein kinase A–mediated serine phosphorylation at amino acid 256 is required for its exocytosis, a process also requiring a heterotrimeric G protein of the G i family. In response to water restriction or desmopressin infusion in humans, the content of urinary AQP2 in both soluble and membrane-bound forms has been found to increase. Mice with targeted deletion of the AQP2 gene have been made. As expected, they have nephrogenic diabetes insipidus that is unresponsive to treatment with vasopressin.
Hypercalciuria is associated with polyuria despite adequate vasopressin levels. The activation by luminal calcium, of the calcium-sensing receptor (CaSR) on the apical membranes of the collecting duct cells of the kidney, is associated with resistance to vasopressin action. The production of dilute urine when there is increased urinary concentration of calcium, has been hypothesized to be a protective mechanism to mitigate the risk of precipitation of calcium in the kidneys. Mechanisms by which the activation of the CaSR decreases AQP2 levels in the collecting duct cells include reduced synthesis of AQP2 by the generation of AQP2 targeting microribonucleic acid (miRNA), and the inactivation and degradation of AQP2 through ubiquitination and phosphorylation at serine261.
In addition to AQP2, different aquaporins appear to be involved in other aspects of renal water handling. In contrast to the apical localization of AQP2, aquaporin-3 and aquaporin-4 are expressed on the basolateral membrane of the collecting duct epithelium. They are involved in the flow of water and urea from the inside of the collecting duct cell into the extracellular renal medullary space. Mice made genetically deficient in aquaporin-4 demonstrate a mild urinary concentrating defect, whereas those with deficiency of aquaporin-3 alone, or together with aquaporin-4, demonstrate more severely impaired urinary concentrating ability. Mice made genetically deficient in aquaporin-1 demonstrate a urinary concentrating defect caused by decreased water permeability in the proximal tubule.
Volume Sensor and Effector Pathways
Renin-Angiotensin-Aldosterone System
In contrast to the vasopressin system, the classic, or peripheral, renin-angiotensin system primarily affects maintenance of intravascular volume as opposed to plasma tonicity. In addition to the well-established endocrine regulatory system, several local renin-angiotensin systems have emerged, with both autocrine and paracrine effects in their tissue of synthesis, whose regulation is independent of the classic system. Finally, brain and pituitary angiotensin systems involved in blood pressure, autonomic function, and fluid balance have been characterized with extensive interaction with the vasopressin system, and vasopressin has been found to play a role in the normal action of aldosterone on the renal tubular epithelium.
Endocrine Renin-Angiotensin-Aldosterone System
Anatomy and Biochemistry
Renin, which is synthesized by the renal juxtaglomerular apparatus, is a proteolytic enzyme that catalyzes the cleavage of angiotensinogen, synthesized by hepatocytes, into the decapeptide angiotensin I. Angiotensin I possesses no intrinsic vasoreactive or mineralocorticoid secretagogue activity, but is efficiently cleaved by angiotensin-converting enzyme in the lungs, as well as other peripheral sites, to generate the octapeptide angiotensin II. Angiotensin II is further metabolized to the heptapeptide angiotensin III by removal of one amino-terminal amino acid. Angiotensin II possesses greater vasopressor activity and is present in approximately a 4-fold greater amount than angiotensin III. Angiotensins II and III possess equivalent mineralocorticoid secretory activity on the adrenal glomerulosa cells.
Angiotensin II and III act through cell surface receptors (AT 1 ) on the adrenal glomerulosa cells to activate the phospholipase C/protein kinase C pathway. This activation results in increased production of pregnenolone from cholesterol by side-chain cleavage enzyme (20,22-desmolase) and of aldosterone from corticosterone by the glomerulosa-specific corticosterone methyloxidase I and II activities (18-hydroxylation and dehydrogenation, respectively). A distinct receptor subtype for angiotensin II and III, the AT 2 receptor, is not G protein coupled and is of unclear physiologic significance in the periphery, but may counteract AT 1 effects by inducing natriuresis. Aldosterone, the primary and most potent endogenous mineralocorticoid released by the zona glomerulosa, acts on target tissues expressing the nuclear mineralocorticoid (or type I glucocorticoid) receptor to promote sodium absorption and potassium excretion. For control of intravascular volume, the primary target of action of aldosterone is the distal nephron. Here, aldosterone increases synthesis of apical membrane sodium channels, mitochondrial enzymes involved in adenosine triphosphate production, and components of Na + , K + adenosine triphosphatase to cause increased sodium reabsorption and potassium excretion.
Regulation of Secretion
Decreased intravascular volume as sensed by the renal juxtaglomerular apparatus results in release of renin. Increased plasma renin activity then allows increased conversion of angiotensinogen to angiotensin I, which in turn is converted peripherally to angiotensins II and III. Increased angiotensin II activity causes vasoconstriction and blood pressure elevation, whereas both angiotensins II and III stimulate aldosterone release from the zona glomerulosa and subsequent salt and water retention and potassium excretion by the distal tubule of the kidney. Conversely, expanded intravascular volume causes decreased renin output and less sodium and water resorption in the kidney, serving to decrease intravascular volume and restore homeostasis.
Changes in vascular volume are not the only regulators of the renin-angiotensin-aldosterone system. Serum potassium concentration directly modulates aldosterone release by the adrenal glomerulosa by its effects on plasma membrane potential and activation of voltage-gated calcium channels. By membrane depolarization, increased serum potassium leads to increased aldosterone synthesis, which promotes renal potassium excretion, whereas low serum potassium reduces aldosterone synthesis and decreases urinary potassium losses. Pituitary adrenocorticotropin hormone and vasopressin act through their respective receptors on the glomerulosa cells to increase acute aldosterone secretion. These effects are of short duration because long-term chronic infusions do not chronically elevate aldosterone concentrations. Direct inhibitors of aldosterone secretion, and thus promoters of natriuresis, include atrial natriuretic peptide (ANP), somatostatin, and dopamine.
Local Renin-Angiotensin Systems
Anatomy and Biochemistry
In addition to the well-defined endocrine circuit, the components of the renin-angiotensin system have been found in a wide variety of tissues, including brain, pituitary, arterial wall, heart, ovary, kidney, and adrenal, where paracrine and autocrine regulatory functions have been postulated, undergoing regulation independent of the systemic counterpart. From the standpoint of regulation of water and volume homeostasis, the brain renin-angiotensin system merits further description. It has long been known that peripherally synthesized angiotensin II could increase blood pressure by effects on the brain outside the blood-brain barrier, at sites such as the OVLT, SFO, area postrema, and median eminence, as revealed by ligand-binding studies. Over the past decade, it has become clear that the complete system for generation of angiotensin II is present within the brain. Angiotensinogen has been localized to astrocytes by both immunohistochemical peptide localization and in situ hybridization analysis of messenger RNA. In contrast, renin has been found in high concentration in nerve terminals, with enhanced release on nerve depolarization. Angiotensin-converting enzyme has been found within vascular, choroid plexus, and neuronal components of the central nervous system, most notably the SFO and many hypothalamic nuclei, sites of endogenous angiotensin II receptor expression, primarily of the AT 1 subtype, as well as sites not expressing the angiotensin II receptor, such as the basal ganglia. The primary effector molecule, angiotensin II, has been localized specifically to neurons and subcellularly to synaptic vesicles. Two of the most significant sites include the circumventricular organs and the paraventricular nucleus of the hypothalamus. Within the paraventricular nucleus, angiotensin II immunoreactivity colocalizes with magnocellular vasopressin, whereas its receptors are within the parvocellular region of the paraventricular nucleus.
Regulation of Secretion
The forebrain angiotensin II pathway, of which the paraventricular nucleus is one component, and circumventricular organ angiotensin II pathway are important control centers for maintenance of osmotic and volume homeostasis. Increased concentration of peripheral angiotensin II, as would be expected in intravascular volume depletion, stimulates drinking behavior. This action of peripheral angiotensin II can be abolished by destruction of the OVLT or SFO, regions whose destruction has long been recognized as causing adipsia. Further effects of central angiotensin II action include augmentation of sodium appetite and stimulation of vasopressin release, all serving, as with peripheral angiotensin II, to restore intravascular volume and maintain blood pressure. The signal of hypovolemia is transduced through the vagal nerve from volume sensors to the brain stem and the region of the nucleus tractus solitarius. Efferents from these brain-stem centers project to the median preoptic nucleus and paraventricular nucleus, as does the forebrain angiotensin II pathway, where drinking and pressor effects, as well as vasopressin release, are elicited.
Separate pathways for vasopressin release mediate the response to either peripheral angiotensin II or purely osmotic stimulation of the osmosensors. The release of vasopressin in response to osmotic stimulation is not increased by peripheral angiotensin II, and pure osmotic stimulation does not increase salt appetite. Central angiotensin II, in contrast, may function as a transmitter in the osmosensing circuit, leading to vasopressin release.
The Natriuretic Peptide System
In addition to the classic vasopressin and renin-angiotensin-aldosterone systems, the natriuretic peptide families of ligands and their receptors add further potential for modulation of salt and water balance. The interaction of the natriuretic peptide system occurs both in the central nervous system through effects on vasopressin secretion, and peripherally, through its ability to both directly promote natriuresis in the kidney and indirectly inhibit adrenal aldosterone production.
Anatomy and Biochemistry
ANP was initially discovered as a component of cardiac atrial muscle that was able to induce natriuresis, a decrease in blood pressure, and an increase in hematocrit when injected into rats.
The biologically active form of ANP consists of a 28-amino-acid peptide that includes a 17-amino-acid ring structure ( Fig. 12.11 ). The primary sequence of the peptide has been conserved among mammalian species and, in addition to synthesis in cardiac atrial tissue, has been detected in brain, spinal cord, pituitary, and adrenal gland. Within the brain, ANP synthesis occurs at several critical neuroendocrine regulatory sites, including the periventricular, arcuate, anteroventral preoptic, and lateral hypothalamic nuclei. ANP is synthesized as a 151-amino-acid preprohormone and is stored as a 126-amino-acid prohormone after removal of the signal peptide sequence. Coupled with secretion of pro-ANP is its cleavage between amino acids 98 and 99 to yield the mature 28-amino-acid 99–126 fragment.

Subsequent investigation defined a second peptide from porcine brain with structural homology to ANP. This peptide, designated brain natriuretic peptide (BNP), was later found to be secreted by the heart as well, in this case, from both ventricular and atrial tissue. Human BNP consists of a 32-amino-acid processed from a larger preprohormone sharing a central ring structure with ANP (see Fig. 12.11 ), although it is less conserved between species than ANP.
A third member of this family, C-type natriuretic peptide (CNP), was also isolated from porcine brain. In brain, CNP is the most abundant member of the natriuretic peptide family. Within the hypothalamus, specific sites of synthesis largely overlap sites of ANP expression. Little CNP can be detected in plasma, and in marked contrast to ANP and BNP, CNP does not increase in plasma in the setting of cardiac failure. Outside the brain, CNP is synthesized in endothelial and vascular smooth muscle. In tissues capable of CNP gene expression, two forms of the peptide are produced, a 53-amino-acid peptide and a less abundant 22-amino-acid molecule (see Fig. 12.11 ).
Three distinct endogenous receptors exist for the natriuretic peptides. The first of these receptors isolated (natriuretic peptide receptor [NPR]-A or guanylyl cyclase [GC]-A) was cloned by virtue of its homology to sea urchin sperm guanylyl cyclase and was later found to have ANP and BNP as its normal ligands. A second guanylyl cyclase type receptor (NPR-B) has substantial homology to NPR-A; however, it binds CNP with substantially greater affinity than ANP or BNP. A third receptor (NPR-C ) does not possess guanylyl cyclase activity and probably functions to clear all three natriuretic peptides from the circulation. In situ hybridization studies using probes capable of distinguishing the different receptor types have revealed some interspecies discrepancy in distribution. The NPR-A receptor has been localized to kidney, adrenal, pituitary, brain, and heart in monkey, with NPR-B limited to adrenal, pituitary, and brain. In rat, broad tissue distribution of both NPR-A and NPR-B has been described. The NPR-C receptor has similarly been found in adrenal, heart, brain, and pituitary.
Regulation of Secretion and Action
Secretion of ANP by cardiac tissue occurs in response to increasing atrial transmural pressure, from both left and right atria. Studies using intravascular volume expansion, exercise, and hypoxia demonstrate increased plasma concentration of ANP after these stimuli in both animal and human paradigms. Also increased heart rate, especially increased atrial contractile frequency, results in increased ANP secretion. In the setting of supraventricular tachycardia, high plasma concentration of ANP, as well as suppressed concentration of vasopressin (both probably caused by an increase in atrial volume and pressure) contribute to the polyuria associated with this syndrome. Ventricular production of ANP has also been demonstrated; it is increased in states of left-sided overload associated with ventricular hypertrophy. ANP synthesized within the central nervous system varies in a volume-dependent fashion, in a manner similar to peripheral ANP, suggesting similar function.
The physiologic ramifications of increased ANP production are several. Infusion of ANP in the setting of normovolemia causes natriuresis, diuresis, and a small increase in divalent cation excretion. ANP, through the NPR-A receptor, primarily inhibits sodium reabsorption within the renal inner medullary collecting duct, but also opposes the salt-retaining effects of angiotensin II at the level of the proximal tubule. ANP similarly inhibits the actions of vasopressin and aldosterone in the renal tubules. Direct cardiovascular effects of ANP include arterial smooth muscle relaxation, both acutely and with chronic administration. In part, this effect may be mediated through opposition of angiotensin II action.
ANP modulates mineralocorticoid production in a manner that results in the reduction of intravascular volume or pressure. Although direct reduction in plasma renin activity has been described with ANP infusion, the most dramatic response to ANP occurs at the level of the adrenal glomerulosa cell. ANP inhibits aldosterone production by inhibiting action of most aldosterone secretagogues, with the most pronounced reduction being angiotensin II activity. The serum concentration of ANP at which the effects on plasma renin activity and aldosterone production occur is within the physiologic range, although the importance of this pathway in normal human physiology remains incompletely defined.
Direct injection of ANP into the central nervous system of animals has suggested an important role for ANP (or CNP) in cardiovascular and salt homeostasis. Hypotension and bradycardia have both been observed, as has inhibition of vasopressin, adrenocorticotropic hormone, and gonadotropin-releasing hormone secretion. Thus antagonistic action of ANP and angiotensin II on intravascular volume and blood pressure remain congruent between central and peripheral systems.
BNP synthesis and secretion from cardiac ventricular tissue are augmented in congestive heart failure, and, as for ANP, with hypertension, chronic renal, and chronic liver failure. BNP binds the NPR-A receptor, where it is capable of stimulating cyclic guanosine monophosphate production. Infusion of BNP inhibits aldosterone production and results in natriuresis similar to that reported for ANP. With infusion rates generating BNP concentrations 10-fold greater than baseline, reduction in blood pressure has also been found. In addition to modulating sodium homeostasis, ANP and BNP, via NPR-A receptors, stimulate the transition of white to beige fat, and therefore may be involved in thermoregulation and energy balance.
In contrast to ANP and BNP, CNP expression primarily causes activation of the NPR-B receptor. Plasma concentration of CNP does not change significantly with volume overload, and it is believed the majority of CNP action occurs in a paracrine fashion, both within the brain and vasculature. CNP synthesized within vascular endothelium acts on receptors in vascular smooth muscle to cause relaxation. CNP infusions in dogs acutely reduce blood pressure and right atrial pressure, but do not result in natriuresis, whereas, in humans, moderately supraphysiologic doses cause neither hypotension nor natriuresis. In contrast to ANP, intracerebroventricular infusion of CNP leads to a reduction in blood pressure, suggesting a role for CNP in central control of arterial pressure. CNP inhibits angiotensin II–stimulated vasopressin secretion but stimulates thirst. The overall importance of the CNP central pathways in modulation of water balance in humans remains to be defined.
Approach to the patient: Differential diagnosis of disorders of water metabolism
Hyponatremia ( Fig. 12.12 )
Hyponatremia (serum sodium < 135 mEq/L) in children is usually associated with severe systemic disorders. It is most often caused by intravascular volume depletion or excessive salt loss and is also encountered with hypotonic fluid overload, especially in infants. Inappropriate vasopressin excess is one of the least common causes of hyponatremia in children, except after vasopressin administration for treatment of diabetes insipidus.
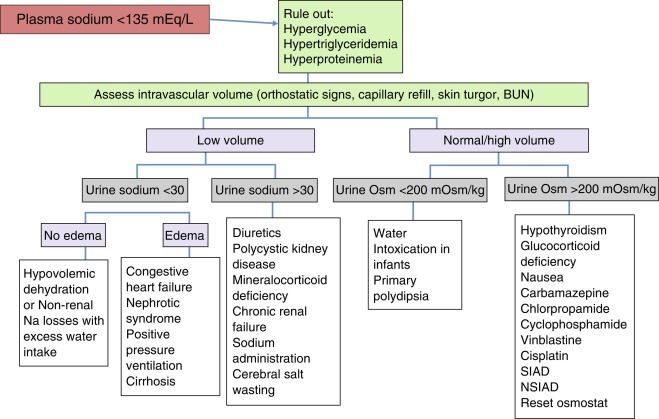
In evaluating the cause of hyponatremia, one should first determine whether the patient is dehydrated and hypovolemic. This is usually evident from the physical examination (decreased weight, skin turgor, central venous pressure) and laboratory data (high blood urea nitrogen, renin, aldosterone, uric acid). With a decrease in the glomerular filtration rate, proximal tubular reabsorption of sodium and water will be high, leading to a urinary sodium value often less than 10 mEq/L. Patients with decreased “effective” intravascular volume from congestive heart failure, cirrhosis, nephrotic syndrome, or lung disease will present with similar laboratory data, but will also have obvious signs of their underlying disease, which often includes peripheral edema. Patients with primary salt loss will also appear volume depleted. If the salt loss is from the kidney (e.g., diuretic therapy or polycystic kidney disease), the urine sodium level will be elevated, as may urine volume. Salt loss from other regions (e.g., the gut in gastroenteritis or the skin in cystic fibrosis) will cause urine sodium to be low, as in other forms of systemic dehydration. Cerebral salt wasting has been invoked to occur with central nervous system insults, and results in high serum ANP concentrations, leading to high urine sodium and urine excretion and systemic dehydration. Its existence is controversial, and believed by many experts to be rare or not exist.
The syndrome of inappropriate antidiuresis (SIAD) exists when a primary elevation in vasopressin secretion or inappropriate activation of the vasopressin V2 receptor is the cause of hyponatremia. It is characterized by hyponatremia, an inappropriately concentrated urine (> 100 mOsm/kg), normal or slightly elevated plasma volume, and a normal-to-high urine sodium (because of volume-induced suppression of aldosterone and elevation of ANP). Serum uric acid is low in patients with SIAD, whereas it is high in those with hyponatremia caused by systemic dehydration or other causes of decreased intravascular volume. Measurement of plasma vasopressin is often not very useful because it is elevated in nearly all causes of hyponatremia, except for primary hypersecretion of ANP or mutations in the vasopressin receptor that lead to its inappropriate activation. Because cortisol and thyroid deficiency cause hyponatremia by several mechanisms, discussed subsequently, they should be considered in all hyponatremic patients. Drug-induced hyponatremia should be considered in patients on potentially offending medications, as discussed later. In children with SIAD who do not have an obvious cause, a careful search for a tumor (thymoma, glioma, bronchial carcinoid) should be considered. Recently, the measurement of copeptin, coupled with hypertonic saline infusion, has been used to classify SIAD into different subtypes.
Polyuria, Polydipsia, and Hypernatremia ( Fig. 12.13 )
In children, it must first be determined whether pathologic polyuria or polydipsia (either exceeding 2 L/m 2 /day) is present. The following questions are asked: Is there a psychosocial reason for either polyuria or polydipsia? Can either be quantitated? Has either polyuria or polydipsia interfered with normal activities? Is nocturia or enuresis present? If so, does the patient also drink after nocturnal awakening? Does the history (including longitudinal growth data) or physical examination suggest other deficient or excessive endocrine secretion or an intracranial neoplasm?
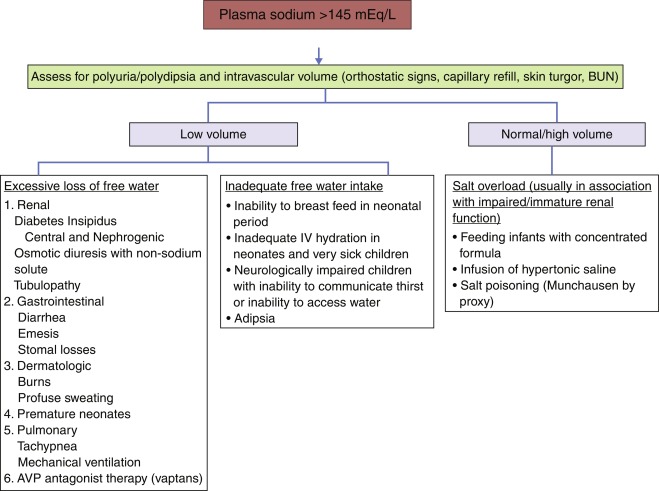
If pathologic polyuria or polydipsia is present, the following should be obtained. In the outpatient setting: serum osmolality; serum concentrations of sodium, potassium, glucose, calcium, and blood urea nitrogen; and urinalysis, including measurement of urine osmolality, specific gravity, and glucose concentration. A serum osmolality greater than 300 mOsm/kg, with urine osmolality less than 300 mOsm/kg, establishes the diagnosis of diabetes insipidus. If serum osmolality is less than 270 mOsm/kg, or urine osmolality is greater than 600 mOsm/kg, the diagnosis of diabetes insipidus is unlikely. If, on initial screening, the patient has a serum osmolality less than 300 mOsm/kg, but the intake/output record at home suggests significant polyuria and polydipsia that cannot be attributed to primary polydipsia (i.e., the serum osmolality is greater than 270 mOsm/kg), the patient should undergo a water deprivation test to establish a diagnosis of diabetes insipidus and to differentiate central from nephrogenic causes.
After a maximally tolerated overnight fast (based on the outpatient history), the child is admitted to the outpatient testing center in the early morning of a day when an 8- to 10-hour test can be carried out, and the child is deprived of water. The physical signs and biochemical parameters shown in the accompanying protocol are measured ( Fig. 12.14 ). If at any time during the test, the urine osmolality exceeds 1000 mOsm/kg, or 600 mOsm/kg and is stable over 1 hour, the patient does not have diabetes insipidus. If at any time the serum osmolality exceeds 300 mOsm/kg and the urine osmolality is less than 600 mOsm/kg, the patient has diabetes insipidus. If the serum osmolality is less than 300 mOsm/kg and the urine osmolality is less than 600 mOsm/kg, the test should be continued unless vital signs disclose hypovolemia. This may be difficult in a young child who may not tolerate a long period of fasting.

A common error is to stop a test too soon, based on the amount of body weight lost, before either urine osmolality has plateaued above 600 mOsm/kg or a serum osmolality above 300 mOsm/kg has been achieved. Unless the serum osmolality increases above the threshold for vasopressin release, a lack of vasopressin action (as inferred by a nonconcentrated urine) cannot be deemed pathologic. As an alternative to water deprivation, hypertonic saline can be infused intravenously to raise serum osmolality above the threshold for vasopressin release. If the diagnosis of diabetes insipidus is made, aqueous vasopressin (Pitressin, 1 U/m 2 ) should be given subcutaneously. If the patient has central diabetes insipidus, urine volume should fall and osmolality should at least double during the next hour, compared with the value before vasopressin therapy. If there is less than a 2-fold increase in urine osmolality after vasopressin administration, the patient probably has nephrogenic diabetes insipidus. Desmopressin should not be used for this test, because it has been associated with water intoxication in small children in this setting. Patients with long-standing primary polydipsia may have mild nephrogenic diabetes insipidus because of dilution of their renal medullary interstitium. This should not be confused with primary nephrogenic diabetes insipidus, because patients with primary polydipsia should have a tendency toward hyponatremia, rather than hypernatremia, in the basal state. Moreover, this form of secondary nephrogenic diabetes insipidus will reverse after cessation of overdrinking. Patients with a family history of X-linked nephrogenic diabetes insipidus, or more rarely, autosomal recessive mutations in the AQP2 water channel (most commonly but not always found in consanguineous families ), can be evaluated for these disorders in the prenatal or perinatal period by DNA sequence analysis, thus allowing therapy to be initiated without delay.
The water deprivation test should be sufficient in most patients to establish the diagnosis of diabetes insipidus and to differentiate central from nephrogenic causes. Plasma vasopressin concentrations may be obtained during the procedure (see Fig. 12.14 ), although they are rarely needed for diagnostic purposes in children. They are particularly helpful in differentiating between partial central diabetes insipidus and nephrogenic diabetes insipidus, in that they are low in the former and high in the latter situation. If urine osmolality concentrates normally, but only after serum osmolality is well above 300 mOsm/kg, the patient may have an altered threshold for vasopressin release, also termed a reset osmostat . This may occur after head trauma, neurosurgery, or brain tumors. More recently, an immunoassay for copeptin, the carboxy-terminus of the vasopressin precursor, has been developed, which may replace the measurement of vasopressin in the evaluation of diabetes insipidus. Copeptin is more stable than vasopressin, and blood concentrations of the two peptides are highly correlated. Copeptin levels correlate well with vasopressin levels and show the same response as vasopressin levels to osmotic and to hemodynamic changes.
The diagnostic accuracy of using a copeptin cut-off value of 4.9 pmol/L (central diabetes insipidus if ≤ 4.9 pmol/L; primary polydipsia if > 4.9 pmol/L), after increasing the plasma sodium level to above 150 mmol/L with a bolus followed by an intravenous (IV) infusion of 3% saline, to differentiate between primary polydipsia, central diabetes insipidus and nephrogenic diabetes insipidus, was found to be significantly higher than that of the indirect water deprivation test (96.5% vs. 76.6%). The hypertonic saline infusion test correctly differentiated between partial diabetes insipidus and primary polyuria in 95.2% of the cases, in contrast to the 73.3% success rate with the use of the indirect water deprivation test in the same study. Nephrogenic diabetes insipidus could be diagnosed without the need for water deprivation, when the baseline copeptin level was greater than 21.4 pmol/L.
Magnetic resonance imaging (MRI) is not very helpful in distinguishing central diabetes insipidus from nephrogenic diabetes insipidus. Normally, the posterior pituitary is seen as an area of enhanced brightness in T1-weighted images after administration of gadolinium. The posterior pituitary “bright spot” is diminished or absent in both forms of diabetes insipidus, presumably because of decreased vasopressin synthesis in central, and increased vasopressin release in nephrogenic disease. In primary polydipsia, the bright spot is normal, probably because vasopressin accumulates in the posterior pituitary during chronic water ingestion, whereas it is decreased in SIAD, presumably because of increased vasopressin secretion. Presence of hyperintensity upon T1-weighted MRI in the pituitary stalk, and its absence in the posterior pituitary region, has been associated with acute postneurosurgical diabetes insipidus. Dynamic, fast-frame, MRI analysis has allowed estimation of blood flow to the posterior pituitary. With this technique, both central and nephrogenic diabetes insipidus are associated with delayed enhancement in the area of the neurohypophysis.
In the inpatient postneurosurgical setting, central diabetes insipidus is likely if hyperosmolality (serum osmolality > 300 mOsm/kg) is associated with urine osmolality less than serum osmolality. One must beware of intraoperative fluid expansion with subsequent hypoosmolar polyuria masquerading as diabetes insipidus. Elevated copeptin levels within 24 hours after neurosurgery, in the hypothalamic-pituitary region, predict the unlikeliness of subsequent central diabetes insipidus, whereas low copeptin levels favor it.
Specific disorders of water metabolism
Hyponatremia With Normal Regulation of Vasopressin
Hyponatremia With Appropriate Decreased Secretion of Vasopressin
Increased Water Ingestion (Primary Polydipsia)
In a hypoosmolar state with vasopressin secretion normally suppressed, the kidney can excrete urine with an osmolality as low as 50 mOsm/kg. Under these conditions, a daily solute load of 500 mOsm/m 2 could be excreted in 10 L/m 2 of urine per day. Neonates cannot dilute their urine to this degree and are prone to develop water intoxication at levels of water ingestion above 4 L/m 2 /day (approximately 60 mL/h in a newborn). This may happen when concentrated infant formula is diluted with excess water, either by accident or in a misguided attempt to make it last longer. A primary increase in thirst, without apparent cause, leading to hyponatremia, has been reported in infants as young 5 weeks of age. In older children, with a normal kidney and the ability to suppress vasopressin secretion, hyponatremia does not occur unless water intake exceeds 10 L/m 2 /day, a feat that is almost impossible to accomplish. Long-standing ingestion of large volumes of water will decrease the hypertonicity within the renal medullary interstitium, which will impair water reabsorption and guard against water intoxication. Hyponatremia will occur at lower rates of water ingestion when renal water clearance is impaired, either because of inappropriately elevated vasopressin secretion or for other reasons.
The rare patient in whom the osmotic thresholds for thirst and vasopressin release are reversed illustrates the importance of the normal relationship between these two responses to osmotic stimulation. If thirst is activated below the threshold for vasopressin release, water intake and hypoosmolality will occur and suppress vasopressin secretion, leading to persistent polydipsia and polyuria. As long as daily fluid intake is less than 10 L/m 2 , hyponatremia will not occur. Despite the presence of polyuria and polydipsia, this entity should not be confused with diabetes insipidus because of the absence of hypernatremia, although desmopressin treatment of such a patient may lower serum osmolality below the threshold for thirst, suppressing water ingestion and the consequent polyuria.
Decreased Renal Free Water Clearance
Adrenal insufficiency, either primary or secondary, has long been known to result in compromised free water excretion. The mechanisms by which glucocorticoids and mineralocorticoids modulate water diuresis have been the subject of substantial investigation. Some studies have demonstrated increased plasma vasopressin activity in the context of glucocorticoid insufficiency, consistent with more recent molecular biologic evidence that glucocorticoids inhibit transcription of the vasopressin gene. Other investigators, however, have failed to detect vasopressin in plasma of patients with adrenal insufficiency and abnormal water clearance. Consistent with vasopressin-independent actions of adrenal steroids on water metabolism, Brattleboro rats with hypothalamic diabetes insipidus manifest impaired excretion of a water load after adrenalectomy. In adrenalectomized Brattleboro rats, glucocorticoid administration restored urine flow rate but did not restore maximal urinary diluting capacity. Conversely, mineralocorticoid administration restored maximal urinary diluting capacity but not flow rate. Thus both mineralocorticoids and glucocorticoids are required for normal free water clearance. In part, these vasopressin-independent actions of mineralocorticoids and glucocorticoids have been attributed to the increased glomerular filtration rate arising from reexpansion of extracellular fluid volume (reduced owing to salt wasting) and improved cardiovascular tone, respectively. By restoring the glomerular filtration rate, more free water is delivered to the distal tubule for excretion. In addition, volume repletion reduces the nonosmotic stimuli for vasopressin release of volume depletion and hypotension. Recently, nitric oxide has been found to stimulate cyclic guanosine monophosphate–dependent membrane insertion of AQP2 into renal epithelial cells. Because glucocorticoid has been shown to inhibit endothelial nitric oxide synthase, it is possible that under conditions of glucocorticoid deficiency, high levels of nitric oxide synthase result in elevated levels of endothelial nitric oxide in the renal vasculature, which in the distal renal tubule stimulate increased, vasopressin-independent, AQP2 activity and decreased free water clearance.
Direct effects of glucocorticoid or mineralocorticoid insufficiency on aquaporin expression and function have not been reported. In addition to impairing maximal renal diluting capacity, adrenal insufficiency compromises maximal urine-concentrating capacity. This effect has been shown to result from reduced tubular response to vasopressin.
Thyroid hormone is also required for normal free water clearance, and its deficiency likewise results in decreased renal water clearance and hyponatremia. Although some studies suggest that vasopressin mediates the hyponatremia of hypothyroidism because ethanol increases free water excretion in hypothyroid patients, this effect has not been found in other reports. In addition, in severe hypothyroidism, hypovolemia is not present and hyponatremia is accompanied by appropriate suppression of vasopressin. Similar to the consequences of isolated glucocorticoid deficiency described earlier, hypothyroidism impairs free water clearance more than maximal urine diluting capacity. This decrease in free water clearance may result from diminished glomerular filtration rate and delivery of free water to the diluting segment of distal nephron, as suggested by both animal and human studies.
Given the often-subtle clinical findings associated with adrenal and thyroid deficiency, all patients with hyponatremia should be suspected of having these disease states and have appropriate diagnostic tests performed if indicated. Moreover, patients with coexisting adrenal failure and diabetes insipidus may have no symptoms of the latter until glucocorticoid therapy unmasks the need for vasopressin replacement. Similarly, resolution of diabetes insipidus in chronically polyuric and polydipsic patients may suggest inadequate glucocorticoid supplementation or noncompliance with glucocorticoid replacement.
Some drugs may cause hyponatremia by inhibiting renal water excretion without stimulating secretion of vasopressin ( Table 12.1 ), an action that could be called nephrogenic SIAD . In addition to augmenting vasopressin release, both carbamazepine and chlorpropamide increase the cellular response to vasopressin. Acetaminophen also increases the response of the kidney to vasopressin; however, this has not been found to cause hyponatremia. High-dose cyclophosphamide treatment (15 to 20 mg/kg IV bolus) is often associated with hyponatremia, particularly when it is followed by a forced water diuresis to prevent hemorrhagic cystitis. Plasma vasopressin concentrations are normal, suggesting a direct effect of the drug to increase water resorption. Similarly, vinblastine, independent of augmentation of plasma vasopressin concentration or vasopressin action, and cisplatin cause hyponatremia. These drugs may damage the collecting duct tubular cells, which are normally highly impermeable to water, or may enhance AQP2 water channel activity and thereby increase water reabsorption down its osmotic gradient into the hypertonic renal interstitium.
| Increases AVP | Increases AVP | AVP-Independent | |||
|---|---|---|---|---|---|
| Class | Drug | Secretion | Effect | Renal Effects | Hyponatremia |
| Angiotensin-converting enzyme inhibitors | Lisinopril | Yes | |||
| Anticonvulsants | Carbamazepine/ oxcarbazepine | Yes | Yes | Possibly | Yes |
| Valproic Acid | Yes | ||||
| Antineoplastics | Cis-platinum | Yes | Yes | ||
| Cyclophosphamide | No | Yes | Yes | ||
| Vinblastine | Yes | Yes | Yes | ||
| Vincristine | Yes | Yes | |||
| Antiparkinsonian | Amantadine, trihexyphenidyl | Yes | Yes | ||
| Antipsychotics | Haloperidol, thioridazine | Yes | |||
| Antipyretics | Acetaminophen | Yes | |||
| Hypolipidemics | Clofibrate | Yes | No | ||
| Oral Hypoglycemics | Chlorpropamide, tolbutamide | Yes | Yes | No | Yes |
| Selective serotonin uptake inhibitors | Fluoxetine, sertraline, others | Likely | Yes | ||
| Tricyclic antidepressants | Imipramine, amitriptyline | Yes | Yes |
Treatment
Hyponatremia caused by cortisol or thyroid hormone deficiency reverses promptly after institution of hormone replacement. Because the hyponatremia is often chronic, too rapid an increase in the serum sodium concentration should be avoided if possible, as will be discussed subsequently. When drugs that impair free water excretion must be used, water intake should be limited, as if the patient has SIAD, to 1 L/m 2 /24 h, using the regimen discussed.
Hyponatremia With Appropriate Increased Secretion of Vasopressin
Increased vasopressin secretion causing hyponatremia may be either an appropriate response or an inappropriate response to a pathologic state. Inappropriate secretion of vasopressin or V2 receptor activity (SIAD) is the much less common of the two entities. Whatever the cause, hyponatremia is a worrisome sign often associated with increased morbidity and mortality.
Systemic Dehydration
Systemic dehydration (water in excess of salt depletion) initially results in hypernatremia, hyperosmolality, and activation of vasopressin secretion, as discussed earlier. In addition, the associated fall in the renal glomerular filtration rate results in an increase in proximal tubular sodium and water reabsorption, with a concomitant decrease in distal tubular water excretion. This limits the ability to form a dilute urine and, along with the associated stimulation of the renin-angiotensin-aldosterone system and suppression of ANP secretion, results in the excretion of urine that is very low in sodium. As dehydration progresses, hypovolemia and/or hypotension become major stimuli for vasopressin release, much more potent than hyperosmolality. This effect, by attempting to preserve volume, decreases free water clearance further and may lead to water retention and hyponatremia, especially if water replacement in excess of salt is given. Other nonosmotic factors may stimulate vasopressin release in a sick child, including pain, nausea, stress and specific conditions, such as pneumonia or meningitis. In many cases, hyponatremia caused by intravascular volume depletion is evident from physical and laboratory signs, such as decreased skin turgor, low central venous pressure, hemoconcentration, and elevated blood urea nitrogen levels. The diagnosis may be subtle, however. For example, patients with meningitis or pneumonia may present with hyponatremia, for which water restriction has been advocated in the belief that it is caused by central SIAD. Several studies have found that volume depletion, rather than SIAD, is often the cause of the hyponatremia, and that it resolves more readily when supplemental, rather than restricted, fluid and solute are administered. In patients with hyponatremia after head trauma, volume depletion rather than central SIAD is the cause in approximately one-half of cases. Similarly, many patients with gastroenteritis who present with mild hyponatremia and elevated plasma vasopressin levels have these on the basis of systemic dehydration rather than SIAD, and benefit from volume expansion rather than fluid restriction. More generally, most hospitalized pediatric patients with hyponatremia benefit from isotonic rather than hypotonic fluid replacement, suggesting that the underlying cause of the electrolyte disturbance is dehydration. Meta-analyses of several studies have revealed that the incidence of hyponatremia in hospitalized children that received hypotonic maintenance IV fluids was significantly higher than those that received isotonic maintenance IV fluids. As a result, the current guidelines for fluid management of hospitalized children recommend the use of isotonic IV fluids except in special clinical situations.
Primary Loss of Sodium Chloride
Salt can be lost from the kidney, such as in patients with congenital polycystic kidney disease, acute interstitial nephritis, and chronic renal failure. Mineralocorticoid deficiency, pseudohypoaldosteronism (sometimes seen in children with urinary tract obstruction or infection), diuretic use, and gastrointestinal disease (usually gastroenteritis with diarrhea and/or vomiting) can also result in excess loss of sodium chloride. Hyponatremia can also result from salt loss in sweat in cystic fibrosis, although obstructive lung disease with elevation of plasma vasopressin probably plays a more prominent role, as has been discussed. With the onset of salt loss, any tendency toward hyponatremia will initially be countered by suppression of vasopressin and increased water excretion. With continuing salt loss, hypovolemia and/or hypotension ensues, causing nonosmotic stimulation of vasopressin. This, plus increased thirst, which leads to ingestion of hypotonic fluids with low solute content, results in hyponatremia. Weight loss is usually evident, as is the source of sodium wasting. If it is the kidney, it is accompanied by a rate of urine output and a urine sodium content greater than those associated with most other causes of hyponatremia, except a primary increase in ANP secretion.
Decreased Effective Plasma Volume
Congestive heart failure, cirrhosis, nephrotic syndrome, positive-pressure mechanical ventilation, severe burns, lung disease (bronchopulmonary dysplasia [in neonates]), cystic fibrosis with obstruction, and severe asthma are all characterized by a decrease in “effective” intravascular volume. This occurs because of impaired cardiac output, an inability to keep fluid within the vascular space, or impaired blood flow into the heart, respectively. As with systemic dehydration, in an attempt to preserve intravascular volume, water and salt excretion by the kidney are reduced; and decreased barosensor stimulation results in a compensatory, appropriate increase in vasopressin secretion, leading to an antidiuretic state and hyponatremia. Because of the associated stimulation of the renin-angiotensin-aldosterone system, these patients also have an increase in the total-body content of sodium chloride and may have peripheral edema, which distinguishes them from those with systemic dehydration. In patients with impaired cardiac output and elevated atrial volume (e.g., congestive heart failure or lung disease), ANP concentrations are elevated, which contributes to hyponatremia by promoting natriuresis.
Treatment
Patients with systemic dehydration and hypovolemia should be rehydrated with salt-containing fluids, such as normal saline or lactated Ringer’s solution. Because of activation of the renin-angiotensin-aldosterone system, the administered sodium will be avidly conserved and a water diuresis will quickly ensue, as volume is restored and vasopressin concentrations fall. Under these conditions, caution must be taken to prevent too rapid a correction of hyponatremia, which may itself result in brain damage.
Hyponatremia caused by a decrease in effective plasma volume from cardiac, hepatic, renal, or pulmonary dysfunction is more difficult to reverse. The most effective therapy is the least easily achieved: treatment of the underlying systemic disorder. Patients weaned from positive-pressure ventilation undergo a prompt water diuresis and resolution of hyponatremia, as cardiac output is restored and vasopressin concentrations fall. The only other effective route is to limit water intake to that required for the renal excretion of the obligate daily solute load of approximately 500 mOsm/m 2 and to replenish insensible losses. In a partial antidiuretic state with a urine osmolality of 750 mOsm/kg H 2 O and insensible losses of 500 mL/m 2 , oral intake would have to be limited to approximately 1200 mL/m 2 /day. Because of concomitant hyperaldosteronism, the dietary restriction of sodium chloride needed to control peripheral edema in patients with heart failure may reduce the daily solute load and further limit the amount of water that can be ingested without exacerbating hyponatremia. Hyponatremia in these settings is often slow to develop, rarely causes symptoms, and usually does not need treatment. If the serum sodium falls below 125 mEq/L, water restriction to 1 L/m 2 /day is usually effective in preventing a further decline. Because water retention in these disorders is a compensatory response to decreased intravascular volume, an attempt to reverse it with drugs, such as demeclocycline or specific V2 receptor antagonists (which induce nephrogenic diabetes insipidus as discussed subsequently) could result in worsening hypovolemia, with potentially dire consequences.
In general, patients with hyponatremia caused by salt loss require ongoing supplementation with sodium chloride and fluids. Initially, IV replacement of urine volume with fluid containing sodium chloride (150 to 450 mEq/L depending on the degree of salt loss) may be necessary; oral salt supplementation may be required subsequently. This treatment contrasts to that of SIAD, in which water restriction without sodium supplementation is the mainstay.
Precautions in the Emergency Treatment of Hyponatremia
Most children with hyponatremia develop the disorder gradually, are asymptomatic, and should be treated with water restriction alone. The development of acute hyponatremia, or a serum sodium concentration below 120 mEq/L, may be associated with lethargy, psychosis, coma, or generalized seizures, especially in younger children. Acute hyponatremia causes cell swelling owing to the entry of water into cells ( Fig. 12.15 ), which can lead to neuronal dysfunction from alterations in the ionic environment or to cerebral herniation because of the encasement of the brain in the cranium. If present for more than 24 hours, cell swelling triggers a compensatory decrease in intracellular organic osmolytes, resulting in the partial restoration of normal cell volume in chronic hyponatremia.
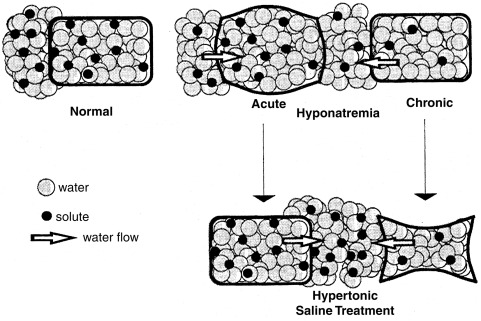
The proper emergency treatment of cerebral dysfunction depends on whether the hyponatremia is acute or chronic. In all cases, water restriction should be instituted. If hyponatremia is acute, and therefore probably not associated with a decrease in intracellular organic osmolyte concentration, rapid correction with hypertonic 3% sodium chloride, administered intravenously, may be indicated. As a general guide, this solution, given in the amount of 12 mL/kg, will result in an increase in serum sodium concentration of approximately 10 mEq/L. If hyponatremia is chronic, hypertonic saline treatment must be undertaken with caution, because it may result in both cell shrinkage (see Fig. 12.15 ) and the associated syndrome of central pontine myelinolysis. This syndrome, affecting the central portion of the basal pons, as well as other brain regions, is characterized by axonal demyelination, with sparing of neurons. It becomes evident within 24 to 48 hours after too rapid correction of hyponatremia, has a characteristic appearance by computed tomography and MRI, and often causes irreversible brain damage. If hypertonic saline treatment is undertaken, the serum sodium concentration should be raised only high enough to cause an improvement in mental status, and in no case faster than 0.5 mEq/L/h or 12 mEq/L/day. In the case of systemic dehydration, the increase in serum sodium level may occur very rapidly using this regimen. The associated hyperaldosteronism will cause avid retention of the administered sodium, leading to rapid restoration of volume and suppression of vasopressin secretion and resulting in a brisk water diuresis and an increase in the serum sodium concentration.
Acute treatment of hyponatremia is more difficult in patients with decreased effective plasma volume. This is both because the underlying disorder makes it difficult to maintain the administered fluid within the intravascular space and because an associated increase in ANP promotes natriuresis and loss of the administered salt. Furthermore, patients with cardiac disease who are administered hypertonic saline may require concomitant treatment with a diuretic, such as furosemide, to prevent worsening of heart failure, which will also increase natriuresis.
Hyponatremia With Abnormal Regulation of Vasopressin
Hyponatremia With Inappropriate Increased Secretion of Vasopressin or Increased Vasopressin V2 Receptor Activity (Syndrome of Inappropriate Antidiuresis)
Causes of SIAD
SIAD is uncommon in children. It can occur with encephalitis, brain tumor, head trauma, brain malformations, in psychiatric disease, and in the postictal period after generalized seizures; after prolonged nausea; pneumonia; or acquired immunodeficiency syndrome. Many drugs have been associated with impaired free water clearance as indicated in Table 12.1 . Impaired free water clearance can result from alteration in vasopressin release, increased vasopressin effect at the same plasma vasopressin concentration, or vasopressin-independent changes in distal collecting tubule water permeability. Common drugs that have been shown to result in hyponatremia include carbamazepine, chlorpropamide, vinblastine, vincristine, and tricyclic antidepressants. Newer sulfonylurea agents, including glyburide, are not associated with SIAD. Other rarer causes of SIAD in children are listed in Table 12.2 . Although it has been believed to be the cause of hyponatremia associated with viral meningitis, volume depletion is more commonly the etiology. In contrast, the majority of children with tuberculous meningitis have hyponatremia and SIAD (in one report attributed to cerebral salt wasting ), which predict more severe disease and poor outcome. SIAD is the cause of the hyponatremic second phase of the triple-phase response seen after hypothalamic-pituitary surgery. Hyponatremia with elevated vasopressin secretion is found in up to 35% of patients 1 week after transsphenoidal pituitary surgery. The mechanism is most likely retrograde neuronal degeneration with cell death and vasopressin release. Secondary adrenal insufficiency causing stimulation of vasopressin release may also play a role, because hyponatremia most commonly follows the removal of adrenocorticotropin hormone-secreting corticotroph adenomas. In the vast majority of children with SIAD, the cause is the excessive administration of vasopressin, whether to treat central diabetes insipidus, or less commonly, bleeding disorders (as has been discussed previously), or, most uncommonly, following dDAVP therapy for enuresis.
| Central Nervous System | Cancer | Infections | Pulmonary |
|---|---|---|---|
| Head trauma | Small cell of lung | Herpes zoster | Viral pneumonia |
| Subarachnoid hemorrhage | Duodenum | Respiratory syncytial virus | Bacterial pneumonia |
| Brain abscess | Pancreas | Tuberculosis | Abscess |
| Guillain-Barre syndrome | Thymoma | Aspergillosis | |
| Hydrocephalus | Bladder | Botulism | |
| Meningitis | Ureter | ||
| Lymphoma | |||
| Ewing sarcoma |
Two unrelated infants with mutations in the vasopressin V2 receptor that presented with severe hyponatremia in the first months of life heralded a new genetic cause of hyponatremia. These two infants had missense mutations at codon 137 that converted arginine to cysteine or leucine and led to constitutive activation of the V2 receptor with appropriately suppressed arginine vasopressin plasma concentration. This genetic disorder has been termed “ nephrogenic syndrome of inappropriate antidiuresis ( NSIAD ).” It remains unclear what portion of isolated early-onset chronic SIAD results from activating mutations of the V2 receptor, although the incidence is likely to be very low. Interestingly, this same codon is also the site of a loss-of-function mutation (R137H), which leads to X-linked nephrogenic diabetes insipidus. Over one dozen patients with NSIAD have been reported, including one patient presenting at the age of 38 years.
Treatment of SIAD
Chronic SIAD is best treated by chronic oral fluid restriction. Under full vasopressin antidiuretic effect (urine osmolality of 1,000 mOsm/L), a normal daily obligate renal solute load of 500 mOsm/m 2 would be excreted in 500 mL/m 2 of water. This, plus a daily nonrenal water loss of 500 mL/m 2 , would require that oral fluid intake be limited to 1000 mL/m 2 /day to avoid hyponatremia, as has been discussed more fully. In young children, this degree of fluid restriction may not provide adequate calories for growth. In this situation, the creation of nephrogenic diabetes insipidus may be indicated to allow sufficient fluid intake for normal growth. Lithium and demeclocycline were previously recommended to induce nephrogenic diabetes insipidus in this situation but are associated with significant toxicities, which limits their use in pediatric patients. Oral urea has been effectively used to treat adult patients with chronic SIAD by virtue of its ability to induce an effective osmotic diuresis. This therapy was also demonstrated to be safe and effective in four children with chronic SIAD, including two with mutations in the vasopressin V2 receptor. Specific nonpeptide V2 receptor antagonists (vaptans) have also been developed for use in subacute or chronic SIAD caused by inappropriate increased vasopressin secretion. Their use is not US Food and Drug Administration (FDA)approved in children. Side effects include increased thirst, dry mouth, elevated hepatic transaminase levels, liver toxicity, and a significant overcorrection of hyponatremia. The aquaretic effects of the vaptans, after either parental or oral administration, have a rapid onset of action, exert peak effects within a few hours, and subside within 24 hours. In a large series of adult patients with euvolemic or hypervolemic hyponatremia resulting from cirrhosis, heart failure, or SIAD, these vasopressin receptor antagonists were effective in sustained elevation of serum sodium concentration. Another study in adults, limited to subjects with SIAD arising from inappropriate vasopressin secretion, demonstrated the efficacy of long-term treatment with an orally active vaptan, in conjunction with fluid restriction to 1.5 L per day. Substantial variability in the degree of serum sodium elevation was observed, however. This variability resulted from both interindividual differences in drug efficacy/disposition, and the failure to adequately restrict water consumption. The primary adverse effect from these agents is inflammation at infusion sites, although rises in serum sodium above rates recommended to prevent myelinolysis have also been found. Very limited experience with vaptans has been reported in children, though they have been used to promote hydration during chemotherapy, with malignancy-associated SIAD. These agents have not been effective in treating activating mutations of the V2 receptor, although low-dose urea has been useful. Because the predictability of hypertonic saline administration for acute, severe forms of SIAD is greater than that of vaptans, hypertonic saline infusion for symptomatic hyponatremia because of inappropriate vasopressin secretion remains the recommended intervention. Of note, some of these V2 receptor antagonists facilitate the proper transport of loss-of-function V2 receptor mutants to the cell surface.
Acute treatment of hyponatremia caused by SIAD is only indicated if cerebral dysfunction is present. In that case, treatment is dictated by the duration of hyponatremia and the extent of cerebral dysfunction. Because patients with SIAD have volume expansion, salt administration is not very effective in raising the serum sodium because it is rapidly excreted in the urine because of suppressed aldosterone and elevated ANP concentrations.
Hyponatremia With Inappropriate Decreased Secretion of Vasopressin, Caused by Increased Secretion of Atrial Natriuretic Peptide
Although ANP does not usually play a primary role in the pathogenesis of disorders of water metabolism, it may have an important secondary role. Patients with SIAD have elevated ANP concentrations, probably caused by hypervolemia, which may contribute to the elevated natriuresis of SIAD and which decrease as water intake is restricted. Likewise, the suppressed ANP concentrations found in central diabetes insipidus, probably caused by the associated hypovolemia, rise after dDAVP therapy. However, hyponatremia in some patients, primarily those with central nervous system disorders, including brain tumor, head trauma, hydrocephalus, neurosurgery, cerebral vascular accidents, and brain death, has been postulated to be caused by the primary hypersecretion of ANP. This syndrome, called cerebral salt wasting , is defined by hyponatremia accompanied by elevated urinary sodium excretion (often more than 150 mEq/L), excessive urine output, hypovolemia, suppressed vasopressin, and elevated ANP concentrations (> 20 pmol/L). Thus it is distinguished from SIAD, in which normal or decreased urine output, euvolemia, only modestly elevated urine sodium concentration, and elevated vasopressin concentration occur. Direct measurement of intravascular volume status with a central venous line is often helpful. The distinction is important because the therapies of the two disorders are markedly different. As noted previously, there is controversy regarding the prevalence of cerebral salt wasting, with most patients actually having either SIAD, dehydration, elevated glucocorticoids, or a combination of these. In patients in an intensive care setting, with the initial diagnosis of cerebral salt wasting, none of them upon further investigation were hypovolemic, one of the cardinal criteria of the syndrome.
Treatment of Cerebral Salt Wasting
Treatment of patients with cerebral salt wasting consists of restoring intravascular volume with sodium chloride and water, as with the treatment of other causes of systemic dehydration. The underlying cause of the disorder, which is usually caused by acute brain injury, should also be treated if possible. A common error is to replace sodium with IV normal saline in the setting of hyperosmolar urine. If urine osmolality is higher than that of the infused saline, hyponatremia may worsen. Instead, saline hypertonic to urine, or enteral sodium chloride, should be used to avoid this issue.
Other Causes of True and Factitious Hyponatremia
True hyponatremia is associated with hyperglycemia, which causes the influx of water into the intravascular space. Serum sodium will decrease by around 1.6 mEq/L for every 100 mg/dL increment in blood glucose above 100 mg/dL. Glucose is not ordinarily an osmotically active agent, and does not stimulate vasopressin release, probably because it is able to equilibrate freely across plasma membranes. However, in the presence of insulin deficiency and hyperglycemia, glucose acts as an osmotic agent, presumably because its normal intracellular access to osmosensor sites is prevented. Under these circumstances, an osmotic gradient exists, and this stimulates vasopressin release. In diabetic ketoacidosis, this, together with the hypovolemia caused by the osmotic diuresis secondary to glycosuria, results in marked stimulation of vasopressin secretion. Rapid correction of hyponatremia may follow soon after the institution of fluid and insulin therapy. Whether this contributes to the pathogenesis of cerebral edema, occasionally seen following treatment of diabetic ketoacidosis, is not known. Elevated concentrations of triglycerides may cause factitious hyponatremia, as can obtaining a blood sample downstream from an IV infusion of hypotonic fluid.
Hypernatremia With Inappropriate Decreased Vasopressin Secretion or Action
Central Diabetes Insipidus
Central (hypothalamic, neurohypophyseal, neurogenic, or vasopressin-sensitive) diabetes insipidus can be caused by disorders of vasopressin gene structure; syndromes that result in loss of vasopressinergic neurons; accidental or surgical trauma to vasopressin neurons; congenital anatomical hypothalamic or pituitary defects; neoplasms; infiltrative, autoimmune, and infectious diseases affecting vasopressin neurons or fiber tracts; and increased metabolism of vasopressin. The etiology of central diabetes insipidus is not apparent in anywhere between 9% and 55% of children and young adults in different series published in the literature. Long-term surveillance can identify an underlying cause that is not apparent at the time of initial diagnosis, sometimes after as long as 21 years.
Genetic Causes
Familial, autosomal dominant central diabetes insipidus is manifest within the first half of the first decade of life. Vasopressin secretion, initially normal, gradually declines until diabetes insipidus of variable severity ensues. Patients respond well to vasopressin replacement therapy. The disease has a high degree of penetrance, but may be of variable severity within a family, and may spontaneously improve in middle age. Vasopressin-containing neurons are absent from the magnocellular paraventricular neurons, but present in parvocellular regions. Several different oligonucleotide mutations in the vasopressin structural gene have been found to cause the disease. To date, there have been over 75 mutations detected in the coding region of the vasopressin gene, with the majority being missense mutations in or around cysteines (see Fig. 12.3 ). Most mutations are in the neurophysin portion of the vasopressin precursor, except for five in either the signal peptide or vasopressin peptide regions of the gene. This suggests that mutant neurophysin has a deleterious function, possibly being misfolded and blocking the proper intracellular sorting or packaging of vasopressin into secretory granules (see later). There are no disease-causing mutations in the copeptin region of the vasopressin precursor. This suggests either that this region has a low mutation rate, that it does not serve a critical function in vasopressin biology, or mutations in it are not disruptive to cellular health.
A family with a missense mutation within the vasopressin peptide region (Proline➔Leucine at amino acid 7) of the gene, causing markedly reduced biological activity, was reported. The disease in this family is transmitted with an autosomal recessive pattern. This indicates that haploinsufficiency is not the basis for the autosomal dominant nature of the disease in families with the more common mutations in the neurophysin region of the gene. Another patient with autosomal recessive central diabetes insipidus presented at a young age (5 months), with compound heterozygous mutations in both copies of AVP, one inherited from each normal parent, again indicating that only one functioning copy is required for normal physiology.
On the other hand, with autosomal dominant familial central diabetes insipidus, the abnormal gene product may interfere with the processing and secretion of the product of the normal allele, or cause neuronal degeneration and cell death. In support of this, mutant vasopressin precursors impair the secretion of the normal protein in cell models, and in a transgenic mouse model of the disease, a progressive loss of hypothalamic vasopressin-containing neurons occurs as the mice develop diabetes insipidus. Heterozygous mice with a mutation (C67X) in the vasopressin gene known to cause autosomal dominant familial neurohypophyseal diabetes insipidus in humans, developed diabetes insipidus by 2 months of age. They were found to have retention of the precursor of vasopressin within the neurons and the induction of an endoplasmic reticulum chaperone protein (BiP). However, humans presenting at less than 1 year of life with central diabetes insipidus are less likely to have autosomal dominant central diabetes insipidus, and more likely to have a structural brain defect.
Vasopressin deficiency is also found in the DIDMOAD syndrome, consisting of diabetes i nsipidus, diabetes mellitus, optic atrophy, and deafness. The gene for this syndrome complex, also known as Wolfram syndrome , was localized to human chromosome 4p16 by polymorphic linkage analysis. The Wolfram syndrome gene, WFS1 , encodes an 890-amino-acid tetrameric transmembrane protein that primarily localizes to the endoplasmic reticulum. It is thought to function as a calcium channel or regulator of a calcium channel. Patients with mutations in proconvertase PCSK1 , required for cleavage of the vasopressin precursor into mature vasopressin and neurophysin, have multiple endocrinopathies including central diabetes insipidus.
Trauma
The axons of vasopressin-containing magnocellular neurons extend uninterrupted to the posterior pituitary over a distance of approximately 10 mm. Trauma to the base of the brain can cause swelling around or severance of these axons, resulting in either transient or permanent diabetes insipidus. Permanent diabetes insipidus can occur after seemingly minor trauma. Approximately one-half of patients with fractures of the sella turcica will develop permanent diabetes insipidus, which may be delayed as long as 1 month following the trauma, during which time neurons of severed axons may undergo retrograde degeneration. Septic shock and postpartum hemorrhage, associated with pituitary infarction (Sheehan syndrome), may involve the posterior pituitary with varying degrees of diabetes insipidus. Diabetes insipidus is never associated with cranial irradiation of the hypothalamic-pituitary region, although this treatment can cause deficits in all of the hypothalamic releasing hormones carried by the portal-hypophyseal system to the anterior pituitary. Because vasopressin is carried directly to the posterior pituitary via magnocellular axonal transport, it may be that radiation affects hypothalamic releasing hormone function by interruption of the portal-hypophyseal circulation, which is absent from the vasopressin circuitry.
Neurosurgical Intervention
One of the most common causes of central diabetes insipidus is the neurosurgical destruction of vasopressin neurons following pituitary-hypothalamic surgery. It is important to distinguish polyuria associated with the onset of acute postsurgical central diabetes insipidus from polyuria caused by the normal diuresis of fluids given during surgery. In both cases, the urine may be very dilute and of high volume, exceeding 200 mL/m 2 /h. However, in the former case, serum osmolality will be high, whereas in the latter case it will be normal. A careful examination of the intraoperative record should also help distinguish between these two possibilities. Vasopressin axons traveling from the hypothalamus to the posterior pituitary terminate at various levels within the stalk and gland (see Fig. 12.4 ). Because surgical interruption of these axons can result in retrograde degeneration of hypothalamic neurons, lesions closer to the hypothalamus will affect more neurons and cause greater permanent loss of hormone secretion. Not infrequently, a “triple phase” response is seen. Although the exact incidence of this phenomenon remains unknown, in a small study, nearly one in three children who underwent surgery for a craniopharyngioma developed it. Following surgery, an initial phase of transient diabetes insipidus is observed, lasting 1.5 to 2 days, and possibly caused by edema in the area interfering with normal vasopressin secretion. If significant vasopressin cell destruction has occurred, this is often followed by a second phase of SIAD, which may last up to 10 days, and is caused by the unregulated release of vasopressin by dying neurons. A third phase of permanent diabetes insipidus may follow if more than 90% of vasopressin cells are destroyed. Usually, a marked degree of SIAD in the second phase portends significant permanent diabetes insipidus in the final phase of this response. In patients with coexisting vasopressin and cortisol deficits (e.g., in combined anterior and posterior hypopituitarism, following neurosurgical treatment of craniopharyngioma), symptoms of diabetes insipidus may be masked because cortisol deficiency impairs renal free water clearance, as discussed subsequently. In such cases, institution of glucocorticoid therapy alone may precipitate polyuria, leading to the diagnosis of diabetes insipidus.
Congenital Anatomic Defects
Midline brain anatomic abnormalities, such as septooptic dysplasia with agenesis of the corpus callosum, the Kabuki syndrome, holoprosencephaly, and familial pituitary hypoplasia with absent stalk may be associated with central diabetes insipidus. These patients need not have external evidence of craniofacial abnormalities. Central diabetes insipidus caused by midline brain abnormalities is often accompanied by defects in thirst perception, suggesting that a common osmosensor may control both vasopressin release and thirst perception. Some patients with suspected defects in osmosensor function but with intact vasopressin neurons may have recumbent diabetes insipidus, with baroreceptor-mediated release of vasopressin, while upright, and vasopressin-deficient polyuria, while supine.
Neoplasms
Several important clinical implications follow from knowledge of the anatomy of the vasopressin system. Because hypothalamic vasopressin neurons are distributed over a large area within the hypothalamus, tumors that cause diabetes insipidus must either be very large or infiltrative or be strategically located at the point of convergence of the hypothalamo-neurohypophyseal axonal tract in the infundibulum. Germinomas and pinealomas typically arise near the base of the hypothalamus where vasopressin axons converge before their entry into the posterior pituitary, and for this reason, are among the most common primary brain tumors associated with diabetes insipidus. Germinomas causing the disease can be very small and undetectable by MRI for several years following the onset of polyuria. For this reason, quantitative measurement of the beta subunit of human chorionic gonadotropin, often secreted by germinomas and pinealomas, and regularly repeated MRI scans should be performed in children with idiopathic or unexplained diabetes insipidus. Empty sella syndrome, possibly caused by unrecognized pituitary infarction, can be associated with diabetes insipidus in children. Craniopharyngiomas and optic gliomas can also cause central diabetes insipidus when very large, although this is more often a postoperative complication of the treatment for these tumors. Hematologic malignancies can cause diabetes insipidus. In some cases, such as with acute myelocytic leukemia, the cause is infiltration of the pituitary stalk and sella. However, more than 30 patients with monosomy or deletion of chromosome 7 associated with acute blast transformation of myelodysplastic syndrome presented with central diabetes insipidus, without evidence of infiltration of the posterior pituitary by neoplastic cells, leaving the cause of the diabetes insipidus unresolved.
Infiltrative, Autoimmune, and Infectious Diseases
Langerhans cell histiocytosis and lymphocytic hypophysitis are the most common types of infiltrative disorders causing central diabetes insipidus. Approximately 10% of patients with histiocytosis will have diabetes insipidus. These patients tend to have more serious, multisystem disease for longer periods of time than those without diabetes insipidus, and anterior pituitary deficits often accompany posterior pituitary deficiency. MRI characteristically shows thickening of the pituitary stalk. One report suggests that in patients with Langerhans cell histiocytosis, radiation treatment to the pituitary region within 14 days of onset of symptoms of diabetes insipidus may result in return of vasopressin function in more than one-third of affected patients.
Lymphocytic infundibuloneurohypophysitis may account for over one-half of patients with “idiopathic” central diabetes insipidus. This entity may be associated with other autoimmune diseases. Image analysis discloses an enlarged pituitary and thickened stalk, and biopsy of the posterior pituitary reveals lymphocytic infiltration of the gland, stalk, and magnocellular hypothalamic nuclei. A necrotizing form of this entity has been described, which also causes anterior pituitary failure and responds to steroid treatment. Diabetes insipidus can also be associated with pulmonary granulomatous diseases, including sarcoidosis. The use of checkpoint inhibitors as anticancer immunotherapy causes an autoimmune hypophysitis that can not only result in anterior pituitary dysfunction, but also central diabetes insipidus, which may be reversible upon discontinuation of the drug.
Whether antibody-mediated destruction of vasopressin cells occurs is controversial. Over one-half of patients with central diabetes insipidus of a nontraumatic cause have antibodies directed against vasopressin-containing cells, and patients with other autoimmune diseases have such antibodies without evidence of diabetes insipidus. Many patients with central diabetes insipidus also have antivasopressin peptide antibodies, although their appearance usually follows institution of vasopressin treatment. It is very possible that antibodies directed against vasopressin-containing cells or vasopressin are not pathogenetic, but instead are markers of prior neuronal cell destruction.
Infections involving the base of the brain, such as meningococcal, Streptococcus pneumoniae , cryptococcal, listeria, toxoplasmosis meningitis, congenital cytomegalovirus infection, congenital syphilis, and nonspecific inflammatory disease of the brain can cause central diabetes insipidus. The disease is often transient, suggesting that it is caused by inflammation rather than destruction of vasopressin-containing neurons.
Brain Death
Central diabetes insipidus can appear in the setting of hypoxic brain death. Although its presence has been suggested as a marker for brain death in children, in some studies, only a minority of patients with brain death manifest the disorder, and up to 15% of patients with cerebral insults and diabetes insipidus ultimately recover brain function. Polyuria in the setting of brain death can be accompanied by high concentrations of plasma vasopressin, suggesting that some cases mistaken for diabetes insipidus are actually caused by other causes, such as cerebral salt wasting with polyuria, as discussed subsequently.
Increased Metabolism of Vasopressin
The metabolic clearance rate of vasopressin increases 4-fold during pregnancy because of the elaboration of a vasopressinase by the placenta. If the mother cannot respond with a concomitant increase in vasopressin action because of preexisting subclinical central or nephrogenic diabetes insipidus, overt, transient disease will appear, usually early in the third trimester (although occasionally earlier ), and resolve within 1 week of delivery. Even without prior defects vasopressin function, an extreme elevation in vasopressinase concentrations in primigravidas with either preeclampsia, liver dysfunction, or multiple gestation, may result in development of the syndrome. The central diabetes insipidus responds to dDAVP, which cannot be metabolized by vasopressinase, and has no oxytocic or hypertensive activity, because it is specific for AVPR2.
Drugs
The most common agent associated with inhibition of vasopressin release and impaired urine concentrating ability is ethanol. Because inhibition of vasopressin release by ethanol can be overcome in the setting of concurrent hypovolemia, clinically important diabetes insipidus caused by ethanol ingestion is uncommon. Phenytoin, opiate antagonists, ketamine, halothane, and alpha-adrenergic agents have also been associated with impaired vasopressin release.
Children With Primary Enuresis
Although normal children have a nocturnal rise in plasma vasopressin, associated with an increase in urine osmolality, and a decrease in urine volume, those with primary enuresis have a blunted or absent rise in vasopressin, and excrete a higher urine volume of lower tonicity. This has suggested that enuretic children have a primary deficiency in vasopressin secretion, although the same outcome could be caused solely by excessive water intake in these children. The use of the V2 agonist dDAVP is highly effective in abolishing bed wetting episodes, although relapse is high once therapy is stopped. Fluid intake must be limited while a child is exposed to the antidiuretic action of dDAVP to guard against water intoxication. It is therefore best that the medication (only available in tablet in the United States) be taken at bedtime, with no further drinking until the morning, after the antidiuretic effect has waned. Nasal spray dDAVP is no longer approved for use in enuretic children because of this safety concern, whereas dDAVP tablets are FDA approved. The efficacy of dDAVP compared with enuretic alarms is similar, as is their relapse rate. Because of safety concerns, enuretic alarms may be preferable.
Treatment of Central Diabetes Insipidus
Fluid Therapy
Patients with otherwise untreated diabetes insipidus crave cold fluids, especially water. With complete central diabetes insipidus, maximum urine concentrating ability is approximately 100 mOsm/kg. Because 5 L of urine would be required to excrete an average daily solute load of 500 mOsm/m 2 , fluid intake must match this to maintain normal plasma tonicity. With an intact thirst mechanism and free access to oral fluids, a person with complete diabetes insipidus can maintain plasma osmolality and sodium in the high normal range, although at great inconvenience. Furthermore, long-standing intake of these volumes of fluid in children can lead to hydroureter, and even hyperfluorosis in communities that provide fluoridated water. With fluid management alone or with the use of medications other than vasopressin or its analogues, these children can develop nonobstructive hydroureteronephrosis, bladder wall thickening and trabeculation, overflow incontinence and impaired renal function requiring a drainage procedure. These complications are more likely to occur in children with nephrogenic, as opposed to central, diabetes insipidus because treatment with vasopressin analogs is used most of the time in children with central diabetes mellitus.
There are two situations in which central diabetes insipidus may be treated solely with high levels of fluid intake, without vasopressin. Vasopressin therapy coupled with excessive fluid intake (usually greater than 1 L/m 2 /day as discussed subsequently) can result in unwanted hyponatremia. Because neonates and young infants receive all of their nutrition in liquid form, the obligatory high oral fluid requirements for this age (3 L/m 2 /day), combined with vasopressin treatment are likely to lead to this dangerous complication. In infants, oral tablet and intranasal liquid desmopressin are not only difficult to administer accurately, but also their use is associated with significant fluctuations in the serum sodium levels. Such neonates may be better managed with fluid therapy alone. A reduced solute load diet will aid in this regard. Human milk is best for this purpose (75 mOsm/kg H 2 O), whereas cow’s milk is worst (230 mOsm/kg H 2 O). For example, in an infant with diabetes insipidus with a fixed urine osmolality of 100 mOsm/kg H 2 O, 300 mL of urine per day is required to excrete the amount of solute consumed in human milk, whereas 900 mL of urine per day is required to excrete the higher amount of solute consumed in cow’s milk. The Similac PM 60/40 formula has a renal solute load of 92 mOsm/kg H 2 O. In addition, supplementation free water may be needed, depending on the severity of the diabetes insipidus. Options, such as 20 to 30 mL of supplemental free water for every 120 to 160 mL of formula, or dilution of the formula with free water have been used. Although children managed with such a regimen may be chronically thirsty, parents may have difficulty keeping up with the voluminous fluid intake and urine output, and poor growth may occur if adequate calories are not provided along with water, these problems are more easily addressed than is life-threatening hyponatremia. Alternatively, thiazide (chlorothiazide, 5–10 mg/kg/dose), twice or thrice daily, and/or amiloride diuretics may be added to facilitate renal proximal tubular sodium and water reabsorption and thereby decrease oral fluid requirements. This therapy may be accompanied by a mild degree of dehydration. More recently, parenteral desmopressin (0.02–0.08 mcg/dose given once or twice daily) has been administered subcutaneously in infants with good results, although this is not approved by the FDA. The successful use of subcutaneous desmopressin in conjunction with a sliding scale fluid regimen has been reported to treat an infant with central diabetes insipidus and an absent thirst mechanism. The desmopressin dose, which was titrated on the basis of plasma sodium levels monitored at home, ranged between 0.01 mcg and 0.024 mcg per dose, given twice daily. Parenteral desmopressin was originally formulated at a concentration of 4 mcg/mL, to be used at a dose of 0.3 mcg/kg/dose to treat bleeding diatheses, such as hemophilia A and von Willebrand disease type 1. Thus care must be taken if it is used at 1/40 to one-fourth of this dose to treat infants with diabetes insipidus. Buccal administration of a diluted intranasal formulation of dDAVP (10 mcg/mL;1–5 mcg twice daily) in infants with central diabetes insipidus, whose mean age was 4.5 months, resulted in normalization and stabilization of sodium levels within days of starting the treatment. Neonates with central diabetes insipidus have been successfully treated with orally administered diluted formulation of intranasal dDAVP (10 mcg/mL) at doses ranging from 2 to 5 mcg twice daily. There was no need to restrict their feed volume, while on this treatment regimen.
In the acute postoperative management of central diabetes insipidus occurring after neurosurgery in children, vasopressin therapy may be successfully used, but extreme caution must be exerted with its use. While under the full antidiuretic effect of vasopressin, a patient will have a urine osmolality of approximately 1000 mOsm/kg and become hyponatremic if she or he receives an excessive amount of fluids, depending on the solute load and nonrenal water losses. With a solute excretion of 500 mOsm/m 2 /day, normal renal function, and nonrenal fluid losses of 500 mL/m 2 /day, fluid intake of greater than 1 L/m 2 /day (two-thirds of the normal maintenance fluid requirement) will result in hyponatremia. In addition, vasopressin therapy will mask the emergence of the SIAD phase of the triple phase neurohypophyseal response to neurosurgical injury (as has been discussed).
Because of the concerns associated with perioperative vasopressin administration, two different approaches to managing central diabetes insipidus in the surgical patient have been used. The first approach may be particularly useful to manage acute postoperative diabetes insipidus in young children. It uses fluids alone and avoids the use of vasopressin. This method consists of matching input and output hourly using between 1 and 3 L/m 2 /day (40–120 mL/m 2 /h). If IV therapy is used, a basal 40 mL/m 2 /h should be given as 5% dextrose (D5) in one-fourth normal saline (normal saline = 0.9% sodium chloride) and the remainder, depending on the urine output, as 5% dextrose in water. Potassium chloride (40 mEq/L) may be added if oral intake is to be delayed for several days. No additional fluid should be administered for hourly urine volumes under 40 mL/m 2 /h. For hourly urine volumes above 40 mL/m 2 /h, the additional volume should be replaced with 5% dextrose up to a total maximum of 120 mL/m 2 /h. For example, in a child with a surface area of 1 m 2 (approximately 30 kg), the basal infusion rate would be 40 mL/h of 5% dextrose in one-fourth normal saline. For an hourly urine output of 60 mL, an additional 20 mL/h 5% dextrose would be given, for a total infusion rate of 60 mL/h. For urine outputs above 120 mL/h, the total infusion rate would be 120 mL/h. In the presence of diabetes insipidus, this will result in a serum sodium in the 150 mEq/L range and a mildly volume contracted state, which will allow one to assess both thirst sensation, as well as the return of normal vasopressin function or the emergence of SIAD. Patients may become mildly hyperglycemic with this regimen, particularly if they are also receiving postoperative glucocorticoids. However, because it does not use vasopressin, this fluid management protocol prevents any chance of hyponatremia.
Vasopressin and Vasopressin Analogs
Evidence suggests that perioperative use of IV vasopressin in children with central diabetes insipidus may be the treatment modality of choice in most situations, resulting in excursions of serum sodium of smaller magnitude and few adverse sequelae. Although IV therapy with synthetic aqueous vasopressin (Pitressin) had been shown to be useful in the management of central diabetes insipidus of acute onset, concern existed as to the safety of its administration in the complex, rapidly changing course of the child recovering from hypothalamic/pituitary surgery. If continuous vasopressin is administered, fluid intake must be limited to 1 L/m 2 /day or two-thirds maintenance fluid administration (assuming normal solute intake and nonrenal water losses as described). The potency of synthetic vasopressin is still measured using a bioassay and is expressed in bioactive units, with one milliunit (mU) equivalent to approximately 2.5 ng of vasopressin. For IV vasopressin therapy, 1.5 mU/kg/h results in a blood vasopressin concentration of approximately 10 pg/mL, twice that needed for full antidiuretic activity. Vasopressin’s effect is maximal within 2 hours of the start of infusion, and one must beware of it sticking to IV bottles and tubing. Occasionally, following hypothalamic (but not transsphenoidal) surgery, higher initial concentrations of vasopressin are required to treat acute diabetes insipidus, which may be attributable to the release of a substance related to vasopressin from the damaged hypothalamo-neurohypophyseal system, which acts as an antagonist to normal vasopressin activity. Much higher rates of vasopressin infusion, resulting in plasma concentrations above 1000 pg/mL, should be avoided, as they may cause cutaneous necrosis, rhabdomyolysis, and cardiac rhythm disturbances.
In light of the considerations described in the previous paragraph, an effective and safe algorithm for management of perioperative central diabetes insipidus with IV infusion of vasopressin has been used with encouraging results. This algorithm starts with the child receiving an IV infusion of normal saline at two-thirds maintenance or 1 L/m 2 /day. A tentative diagnosis of intraoperative or postoperative central diabetes insipidus is made by documenting a serum sodium of greater than 145 mEq/L, along with urine output of greater than 4 mL/kg/h. Additional confirmatory evidence includes plasma osmolarity above 300 mOsm/kg H 2 O and relatively hypotonic urine. When documentation of parameters consistent with central diabetes insipidus is obtained, an IV infusion of aqueous vasopressin begins at 0.5 mU/kg/h, with no change in IV fluid administration. The dose of vasopressin is titrated upward in 0.5 mU/kg/h increments to establish a urine output rate of less than 2 mL/kg/h at approximately 10-minute intervals. The vasopressin and IV fluid administration then remain stable at these rates, with additional normal saline, or equivalent volume expanding solutions, given only to replace ongoing blood loss or to maintain hemodynamic stability. Postoperatively, this management paradigm requires intensive care unit monitoring, with frequent assessment of electrolytes (hourly initially), urine output and osmolarity/specific gravity, and vital signs. This management scheme can also be followed for patients with established central diabetes insipidus requiring general surgery and prolonged restriction of oral intake. In this situation, the usual dose of chronic, long-acting vasopressin is withheld or reduced immediately before surgery, depending upon the timing of surgery in relation to the usual administration times. In preparation for surgery, normal saline is infused at two-thirds maintenance (1 L/m 2 /day). When measures consistent with emergence of central diabetes insipidus caused by termination of efficacy of the presurgical dose are obtained, the IV vasopressin is initiated and titrated as described earlier.
Patients treated with vasopressin for postneurosurgical diabetes insipidus should be switched from IV to oral fluid intake at the earliest opportunity, because thirst sensation, if intact, will help regulate blood osmolality, as discussed. With the concomitant absence of vasopressin secretion and thirst, the clinician must perform administration fluids and an antidiuretic agent. This must be done with utmost care, for the therapeutic window for fluid administration is narrow when the patient cannot modulate their own antidiuretic activity. Either the patient can be put into a persistent antidiuretic state, with dDAVP or vasopressin, and fluid strictly limited to 1 L/m 2 /day, or the patient can be given intermittent dDAVP with clear urinary “breakthrough” between doses, and a fixed amount of fluid empirically determined to result in normal blood electrolyte concentrations. IV dDAVP (desmopressin) should not be used in the acute management of postoperative central diabetes insipidus, for it offers no advantage over vasopressin, and its long half-life (8–12 hours) compared with that of vasopressin (5–10 minutes) is a distinct disadvantage, as it may increase the chance of causing water intoxication. In fact, the use of IV dDAVP, 0.3 mcg/kg, to shorten the bleeding time in a variety of bleeding disorders (as has been discussed), has been associated with water intoxication, particularly in young children who have high obligate oral fluid needs.
A special problem arises when a patient with established central diabetes insipidus must receive a high volume of fluid for therapeutic reasons, for example, accompanying cancer chemotherapy. Such patients can be managed by discontinuing antidiuretic therapy and increasing fluid intake to 3 to 5 L/m 2 /day (rendering the patient moderately hypernatremic). This would be similar to what would occur in a patient with intact vasopressin secretion, in whom overhydration to this extent would suppress endogenous vasopressin secretion. However, although 5 L/m 2 /day is typically adequate to maintain serum sodium concentration in the range of 150 mEq/L in children with central diabetes insipidus, this rate may not be adequate in the setting of chemotherapy administration when solute excretion increases because of cell death and release of cellular contents. By using a low dose of IV vasopressin (0.08–0.1 mU/kg/h, approximately one-eighth the full antidiuretic dose, titrated upward as needed), a partial antidiuretic effect allows the administration of higher amounts of fluid without causing hyponatremia. Whereas management of diabetes insipidus, with fluids alone, may be used for chemotherapy with ifosfamide (which is associated with nephrotoxicity and hemorrhagic cystitis), recent data suggest that excretion of a hypotonic urine, as would occur in patients with diabetes insipidus management with fluids alone, increases the risk of developing nephrotoxicity during therapy with platinum-based antineoplastic agents. By allowing administration of 0.45% saline at rates of approximately 3 L/m 2 /day, the low-dose vasopressin infusion yields a urine osmolarity higher than that achievable with fluids alone, and may have the additional benefit of conferring renal protection.
In the outpatient setting, treatment of central diabetes insipidus in older children should begin with oral (see later) or intranasal dDAVP (10 mcg/0.1 mL), 0.025 mL (2.5 mcg) given by rhinal tube at bedtime and the dose increased to the lowest amount that gives an antidiuretic effect. If the dose is effective, but has too short a duration, it should be increased further, or a second, morning dose should be added. Patients should escape from the antidiuretic effect for at least 1 hour before the next dose, to ensure that any excessive water will be excreted. Otherwise, water intoxication may occur. dDAVP is also available as a nasal spray in the same concentration, with each spray delivering 10 mcg (0.1 mL). This is the standard preparation that was used for treatment of primary enuresis before this indication was removed. Oral dDAVP tablets and dissolvable wafers are now in widespread use and have largely replaced intranasal therapy. Although, when given orally, dDAVP is at least 20-fold less potent than when given via the intranasal route, oral dDAVP in doses of 25 to 300 mcg every 8 to 12 hours is reported to be highly effective and safe in children. Lysine vasopressin (Diapid) nasal spray (50 units/mL) may be used if a duration less than that of dDAVP is desired. One spray delivers 2 units (0.04 mL), with a duration of action between 2 and 8 hours.
As noted previously, cortisol deficiency may cause decreased free water clearance by stimulating a nitric oxide-mediated pathway, which results in the insertion of AQP2 channels into the apical membranes of collecting duct cells, in a vasopressin-independent fashion. Conversely, it is possible that excessive amounts of cortisol, caused by endogenous release during stress, or to treatment with exogenous drug, may inhibit the insertion of water channels. This may explain why patients, with central diabetes insipidus treated with desmopressin, become “resistant,” and require an increased dosage, during times of stress or treatment with glucocorticoids.
In addition to polyuria and polydipsia, decreased bone mineral density has been reported in patients with central diabetes insipidus. The decreased bone density was not corrected by vasopressin analog treatment alone, suggesting that institution of bisphosphonate or other therapies designed to prevent bone loss may be of long-term benefit in the treatment of diabetes insipidus.
Adipsic Hypernatremia
A subset of patients with central diabetes insipidus also have disorders of thirst mechanism. Lesions that affect the sensory circumventricular organs of the hypothalamus, including the organum vasculosum of the lamina terminalis, subfornical organ, and the area postrema, can result in either absent thirst or an elevated osmotic threshold for thirst perception. These areas lie outside the blood-brain barrier and constantly measure osmolality and sodium levels. Patients with diabetes insipidus and an intact thirst mechanism rarely have a significant hypernatremia because the thirst mechanism ensures that their water intake matches their excessive urinary water losses. However, patients with diabetes insipidus and adipsia can have severe hypernatremia and complications, such as rhabdomyolysis, acute renal failure, and venous thrombosis. Etiologies for adipsic hypernatremia include congenital malformations, such as holoprosencephaly and septooptic dysplasia sequence; vascular lesions, such as anterior communicating artery aneurysm; neoplasms, such as craniopharyngiomas and suprasellar germinomas; granulomatous conditions, such as histiocytosis and sarcoidosis; and miscellaneous conditions, such as arachnoidal cysts and hydrocephalus. Recently, there has been a case series reported of adipsic hypernatremia, without any abnormality on MRI of the hypothalamus and the pituitary gland. The affected children had associated hyperprolactinemia, childhood obesity, and impaired growth hormone secretion. The presence of antibodies targeting the subfornical organ in these patients strongly suggests an autoimmune etiology for their adipsic hypernatremia. The treatment of adipsic hypernatremia involves giving a fixed daily dose of an dDAVP with controlled daily fluid intake and strict monitoring of fluid balance, body weight, and plasma sodium levels. Successful treatment of an infant with diabetes insipidus and absent thirst has been reported using a sliding scale fluid regimen and subcutaneous dDAVP, guided by the frequent home monitoring of plasma sodium levels.
Nephrogenic Diabetes Insipidus
Causes of Nephrogenic Diabetes Insipidus
Nephrogenic (vasopressin-resistant) diabetes insipidus can be caused by genetic or acquired causes. Genetic causes are less common but more severe than acquired forms of the disease, although genetic etiologies are more common in children than in adults.
Genetic Causes
Congenital, X-Linked Diabetes Insipidus: V2 Receptor Mutations
Congenital, X-linked nephrogenic diabetes insipidus is caused by inactivating mutations of the vasopressin V2 receptor. Approximately 250 mutations, mostly missense, have been reported to be associated with nephrogenic diabetes insipidus. Because of its mode of transmission, it is a disease of males, although rarely females may be affected, presumably caused by extreme lyonization during X chromosome inactivation. In keeping with a germline, as opposed to somatic mutation in the V2 receptor, these patients are deficient in all systemic V2 receptor-mediated actions, and have intact V1-receptor–mediated responses. As expected, the V2 receptor defect is proximal to the activation of renal adenylate cyclase. Unlike the function of other G protein–coupled seven transmembrane receptors, such as the parathyroid hormone and thyroid-stimulating hormone receptors, that of the V2 receptor is unaffected in patients with pseudohypoparathyroidism, who have inactivating mutations in the alpha subunit of G s.
Because of vasopressin resistance in congenital nephrogenic diabetes insipidus, the kidney elaborates large volumes of hypotonic urine, with osmolality ranging between 50 and 100 mOsm/kg. Manifestations of the disease are usually present within the first several weeks of life, but may only become apparent after weaning from the breast. The predominant symptoms are polyuria and polydipsia. Thirst may be more difficult to satisfy than in central diabetes insipidus. Many infants initially present with fever, vomiting, and dehydration, often leading to an evaluation for infection. Growth failure in the untreated child may be secondary to the ingestion of large amounts of water, which the child may prefer over milk and other higher caloric substances. Mental retardation of variable severity may result from repeated episodes of dehydration. Intracerebral calcification of the frontal lobes and basal ganglia is not uncommon in children with X-linked nephrogenic diabetes insipidus. Because this appears early and is not seen in children with central diabetes insipidus of equivalent severity, cerebral calcification is probably unrelated to the level of dehydration or therapeutic intervention. It is possible that elevated vasopressin concentrations, acting via intact V1 or V3 receptors, contribute to some of the unique manifestations of X-linked nephrogenic diabetes insipidus, such as cerebral calcification, intense thirst, vomiting, and growth failure. Older children may present with enuresis or nocturia. They may learn to reduce food intake (and therefore solute load) to decrease polyuria, which may contribute to growth failure. After long-standing ingestion and excretion of large volumes of water, patients may develop nonobstructive hydronephrosis, hydroureter, and megabladder.
Although one founder (arriving in North America from Scotland in 1761 on the ship Hopewell ) was initially postulated to be the ancestor of most North American subjects with congenital, X-linked nephrogenic diabetes insipidus, hundreds of independent mutations in the V2 receptor have been found, with some appearing to have arisen independently more than once ( Fig. 12.16 ). These are mostly single base mutations that either result in amino acid substitutions, translational frameshifts, or termination of peptide synthesis, and are distributed fairly evenly throughout the receptor protein. Mutations may affect either vasopressin binding, cyclic AMP generation, or possibly transcriptional regulation. Patients with different mutations will likely be found to exhibit phenotypic heterogeneity, including in severity of disease and response to treatment. Genetic heterogeneity may underlie the variable response of patients with X-linked diabetes insipidus to dDAVP treatment. In a family with a known mutation, prenatal or early postnatal DNA screening can unambiguously identify affected males, allowing the institution of appropriate therapy.