Abbreviations
- ABCA1 ATP-binding cassette transporter A1
- ABCG1 ATP-binding cassette transporter G1
- ACAT Acyl-CoA:cholesterol acyltransferase
- Apo Apolipoprotein
- ARH Autosomal recessive hypercholesterolemia
- CETP Cholesteryl ester transfer protein
- CHD Coronary heart disease
- CK Creatine kinase
- FCH Familial combined hyperlipidemia
- FFA Free fatty acids
- FH Familial hypercholesterolemia
- GPIHDLBP Glycosylphosphatidylinositol-anchored HDL-binding protein
- HDL High-density lipoproteins
- HMG-CoA Hydroxymethylglutaryl-CoA
- IDL intermediate density lipoproteins
- Idol Inducible degrader of the LDL receptor
- LCAT Lecithin-cholesterol acyltransferase
- LDL Low-density lipoproteins
- LMF1 Lipase maturation factor 1
- Lp(a) Lipoprotein(a)
- LPL Lipoprotein lipase
- LRP-1 LDL receptor-related protein-1
- MCP-1 Monocyte chemoattractant protein-1
- NAFLD Nonalcoholic fatty liver disease
- NASH Nonalcoholic steatohepatitis
- PCPE-2 Procollagen C-proteinase enhancer-2
- PCSK9 Proprotein convertase subtilisin/kexin type 9
- PDGF Platelet-derived growth factor
- SR-BI Scavenger receptor, class B, type I
- VLDL Very low density lipoproteins
Disorders of Lipoprotein Metabolism: Introduction
The clinical importance of lipoprotein disorders derives chiefly from the role of lipoproteins in atherogenesis and its associated risk of coronary and peripheral vascular disease. The greatly increased risk of acute pancreatitis associated with severe hypertriglyceridemia is an additional indication for intervention. Disordered lipid metabolism is also a critical element in nonalcoholic fatty liver disease. Characterization of dyslipidemia is important for selection of appropriate treatment and may provide clues to underlying primary clinical disorders.
Atherosclerosis is the leading cause of death in the United States. Abundant epidemiologic evidence establishes its multifactorial character and indicates that the effects of the multiple risk factors are at least additive. Risk factors include hyperlipidemia, hypertension, smoking, diabetes, physical inactivity, decreased levels of high-density lipoproteins (HDL), hyperhomocysteinemia, and hypercoagulable states. Atheromas are complex lesions containing cellular elements, collagen, and lipids. The progression of the lesion is chiefly attributable to its content of unesterified cholesterol and cholesteryl esters. Cholesterol in the atheroma originates in circulating lipoproteins. Atherogenic lipoproteins include low-density (LDL), intermediate density (IDL), very low density lipoproteins (VLDL), and Lp(a) species, all of which contain the B-100 apolipoprotein (Apo B-100). Chylomicron remnants containing apoB-48 are also atherogenic. All of these are subject to oxidation by reactive oxygen species in the tissues and also by lipoxygenases secreted by macrophages in atheromas. Oxidized lipoproteins cause impairment of endothelial cell-mediated vasodilation and stimulate endothelium to secrete monocyte chemoattractant protein-1 (MCP-1) and adhesion molecules that recruit monocytes to the lesion. Tocopherols (vitamin E) are natural antioxidants that localize in the surface monolayers of lipoproteins, exerting resistance to oxidation. Increased oxidative stress such as that induced by smoking depletes the tocopherol content. Oxidation of lipoproteins stimulates their endocytosis via scavenger receptors on macrophages and smooth muscle cells, leading to the formation of foam cells. Recent studies strongly support a role of vitamin D in prevention of atherosclerosis, probably by influencing inflammatory activity of macrophages.
Hypertension increases access of lipoproteins to the subintima. Smoking accelerates atherogenesis by reducing HDL and increasing thrombogenesis by platelets—in addition to its pro-oxidant effect. Activated platelets release platelet-derived growth factor (PDGF), stimulating proliferation and migration of cells of smooth muscle origin into the lesion.
Activated macrophages secrete cytokines that drive an inflammatory and proliferative process. Metalloproteases secreted by macrophages weaken the atheroma so that fissuring and rupture can occur. Exposure of blood to subintimal collagen and tissue factor stimulates thrombogenesis, precipitating acute coronary events. The inverse relationship between HDL levels and atherogenesis probably reflects the role of certain species of HDL in cholesterol retrieval and in protecting lipoproteins against oxidation.
Angiographic intervention trials have shown that regression of atherosclerotic lesions can occur with lipid-lowering therapy. Large trials have demonstrated striking reductions in the incidence of new coronary events in individuals with hyperlipidemia who have had no prior clinical coronary disease (primary prevention) as well as in patients with antecedent disease (secondary prevention). Thus, timely hypolipidemic therapy appropriate to the lipid disorder decreases the incidence of coronary disease and reduces the need for angioplasty, stenting, and bypass surgery. Side-effects of treatment have been minimal in comparison with the magnitude of this benefit.
Average levels of LDL in the United States and northern Europe are higher than in many other nations, where the levels appear to approach the biologic norm. This probably accounts in large part for the higher incidence of coronary disease in industrialized Western nations and suggests that dietary changes that reduce lipoprotein levels would be beneficial.
Because lipids are relatively insoluble in water, they are transported in association with proteins. The simplest complexes are those formed between unesterified, or free, fatty acids (FFA) and albumin, which serve to carry the FFA from peripheral adipocytes to other tissues.
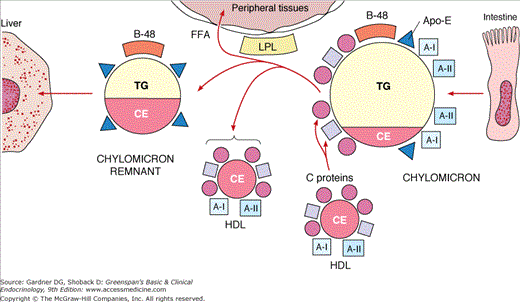
The remainder of the lipids are transported in spherical lipoprotein complexes (Table 19–1), with core regions containing hydrophobic lipids. The principal core lipids are cholesteryl esters and triglycerides. Triglycerides predominate in the cores of chylomicrons, which transport newly absorbed lipids from the intestine, and in VLDL, which originate in liver. The relative content of cholesteryl ester is increased in the cores of remnants derived from these lipoproteins. Cholesteryl esters are the predominant core lipid in LDL and HDL. Surrounding the core in each lipoprotein is a monolayer containing amphiphilic phospholipids and unesterified (free) cholesterol. Apolipoproteins, noncovalently bound to the lipids, are located on this surface monolayer (Figure 19–1).
| Density Interval (g/cm3) | Core Lipids | Diameter (nm) | Apolipoproteins in Order of Quantitative Importance | |
|---|---|---|---|---|
| High density (HDL) | 1.21-1.063 | Cholesteryl ester | 7.5-10.5 | A-I, A-II, C, E (many others) |
| Low density (LDL) | 1.063-1.019 | Cholesteryl ester | 21.5 | B-100 |
| Intermediate density (IDL) | 1.019-1.006 | Cholesteryl ester, triglyceride | 25-30 | B-100, some C and E |
| Very low density (VLDL) | <1.006 | Triglyceride cholesteryl ester | 39-100 | B-100, C, E |
| Chylomicrons | <1.006 | Triglyceride | 60-500 | B-48, C, E, A-I, A-II, A-IV |
| Lp(a) | 1.04-1.08 | Cholesteryl ester | 21-30 | B-100, (a) |
Several lipoproteins contain very high molecular weight B proteins that behave like intrinsic proteins of cell membranes. Unlike the smaller apolipoproteins, the B proteins do not migrate from one lipoprotein particle to another. VLDL contain the B-100 protein, which is retained in the formation of LDL from VLDL remnants by liver. The intestinal B protein, B-48, is found only in chylomicrons and their remnants. Apo B-100 has a ligand domain for binding to the LDL receptor that is conformed as VLDL are transformed into LDL.
In addition to the B proteins, the following proteins are present in lipoproteins (see Table 19–1.)
C apolipoproteins are lower molecular weight proteins that equilibrate rapidly among the lipoproteins. There are four distinct species: C-I, C-II, C-III, and C-IV. Apo C-II is a requisite cofactor for lipoprotein lipase.
Three isoforms of Apo E—E-2, E-3, and E-4—are the products of allelic genes. Unlike Apo E-3 and Apo E-4, Apo E-2 does not contain a functional ligand for the LDL receptor. The Apo E-4 alleles are associated with early-onset Alzheimer disease and increased risk of coronary disease.
Apo A-I is the major apolipoprotein of HDL. It is also present in chylomicrons and is the most abundant of the apolipoproteins of human serum (about 125 mg/dL). It is a cofactor for lecithin-cholesterol acyltransferase (LCAT).
Apo A-II is an important constituent of HDL. It contains cysteine, which permits the formation of disulfide-bridged dimers with Apo E.
Apo A-IV is chiefly associated with chylomicrons.
Apo A-V plays a significant role in the removal of triglycerides from plasma.
The (a) protein is a glycoprotein that has a high degree of sequence homology with plasminogen, resulting in binding affinity for tissue plasminogen activator. It is found as a disulfide-bridged dimer with Apo B-100 in LDL-like species of lipoproteins (Lp[a] lipoproteins).
Dietary triglycerides are hydrolyzed in the intestine to β-monoglyceride and fatty acids by pancreatic lipase, which is activated by bile acids and a protein cofactor. The partial glycerides and fatty acids form micelles that are absorbed by intestinal epithelium. The fatty acids are reesterified with β-monoglycerides to form triglycerides, and free cholesterol is esterified with fatty acids by acyl-CoA:cholesterol acyltransferase-2 (ACAT-2). Droplets of triglyceride with small amounts of cholesteryl esters, associated with B-48, acquire a monolayer of phospholipid and free cholesterol. Apo A-I and Apo A-II are added, and the nascent chylomicron emerges into the extracellular lymph space (see Figure 19–1). The new chylomicron begins to exchange surface components with HDL, acquiring Apo C and Apo E and losing phospholipids. This process continues as the chylomicron is carried via the intestinal lymphatics to the thoracic duct and thence into the bloodstream.
The liver exports triglycerides to peripheral tissues in the cores of VLDL (Figure 19–2). These triglycerides are synthesized in liver from free fatty acids abstracted from plasma and from fatty acids synthesized de novo. Release of VLDL by liver is augmented by any condition that results in increased flux of FFA to liver in the absence of compensating ketogenesis. Obesity, increased caloric intake, ingestion of ethanol, and estrogens stimulate release of VLDL and are important factors in hypertriglyceridemia.
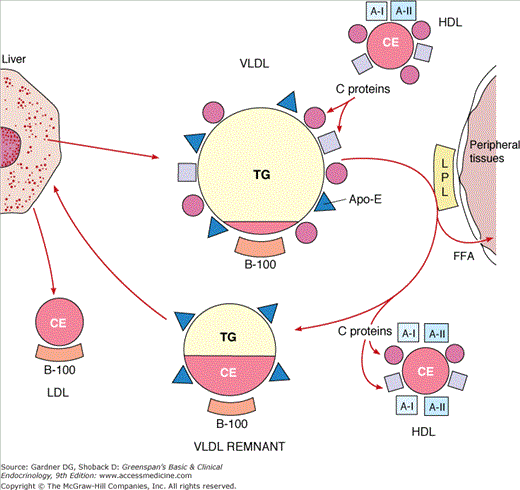
Fatty acids derived from the triglycerides of chylomicrons and VLDL are delivered to tissues through a common pathway involving hydrolysis by the lipoprotein lipase (LPL) system. Members of a family of fatty acid transfer proteins facilitate FFA uptake in tissues. LPL is bound to capillary endothelium in heart, skeletal muscle, adipose tissue, mammary gland, and other tissues.
When glucose levels in plasma are elevated and the release of insulin is stimulated, LPL is transcriptionally upregulated in adipose tissue, and fatty acids derived from triglycerides of circulating lipoproteins are stored. During prolonged fasting, and in diabetic ketoacidosis, LPL activity of adipose tissue falls, preventing storage of fatty acids. Heparin is a cofactor for LPL. When it is given intravenously (0.1-0.2 mg/kg), LPL activity is displaced into plasma, permitting its measurement. Apo C-II is an obligatory cofactor.
Hydrolysis by LPL results in depletion of triglycerides in the cores of chylomicrons and VLDL, producing progressive decreases in particle diameter. Lipids from the surface and C proteins are transferred to HDL. The remnant lipoproteins thus formed contain their original complement of Apo B, significant amounts of Apo E, and little Apo C. They have lost about 70% of their triglycerides and are enriched in cholesteryl esters.
Chylomicron remnants are removed from blood quantitatively by high-affinity, receptor-mediated endocytosis in the liver. The receptors include the LDL (B-100:E) receptors and the LDL receptor-related protein-1 (LRP-1). Endocytosis of chylomicron remnants by both requires the presence of Apo E-3 or Apo E-4. The lipids enter hepatic pools, and B-48 protein is degraded. Cholesterol derived from chylomicron remnants exerts feedback control of cholesterol biosynthesis in liver. Some VLDL remnants are removed from blood via the B-100:E receptors (see later) and are degraded. Those that escape uptake are transformed into LDL. Thus, the rate of removal of VLDL remnants is a determinant of LDL production. Formation of LDL involves the removal of residual triglycerides by hepatic lipase, facilitated by Apo E. LDL contain cholesteryl esters in their cores and retain Apo B-100. In normal individuals, a major fraction of VLDL is converted to LDL, and all of the LDL Apo B comes from VLDL. In certain hypertriglyceridemic states, conversion of VLDL to LDL is decreased. In the absence of impaired conversion, increased secretion of VLDL results in increased production of LDL. This precursor-product relationship explains the clinical phenomenon referred to as the beta shift, an increase of LDL (beta-lipoprotein) as hypertriglyceridemia resolves. An example of this occurs temporarily following institution of insulin treatment in uncontrolled diabetes with lipemia. Insulin induces LPL activity, resulting in rapid conversion of VLDL to LDL. Because of its longer half-life, LDL accumulates in plasma. Elevated levels of LDL may persist beyond the time when levels of triglyceride-rich lipoproteins have returned to normal. A similar phenomenon may occur when patients with familial combined hyperlipidemia (FCH) are treated with fibric acid derivatives.
Normally, the half-life of chylomicrons is 5 to 20 minutes; that of VLDL is 0.5 to 1 hour; and that of LDL is about 2½ days. At triglyceride levels of 800 to 1000 mg/ dL, LPL is at kinetic saturation. Increased input of triglycerides into plasma at those levels rapidly augments the hypertriglyceridemia.
Individuals consuming a typical North American diet transport 75 to 100 g or more of triglyceride per day in chylomicrons, whereas the liver exports 10 to 30 g in VLDL. When LPL is saturated and triglycerides are measured in thousands of milligrams per deciliter, acute restriction of dietary fat produces a significant reduction in levels. This intervention is especially important in the lipemic patient with impending pancreatitis. If symptoms suggest that pancreatitis is imminent, oral intake should be eliminated, gastric acid should be suppressed with H2 blockade, and the patient should not be fed by mouth until the symptoms subside and triglycerides decrease to less than 800 to 1000 mg/dL.
LDL catabolism is mediated by high-affinity receptors on the cell membranes of virtually all nucleated cells but most importantly hepatocytes. Ligands for these LDL receptors exist in Apo B-100 and Apo E. After endocytosis, Apo B is degraded, and the receptor returns to the cell membrane. The cholesteryl esters of LDL are hydrolyzed to free cholesterol for production of cell membrane bilayers. Free cholesterol down regulates hydroxymethylglutaryl-CoA (HMG-CoA) reductase and other rate-limiting enzymes in the biosynthetic pathway for cholesterol. Cholesterol in excess of need for membrane synthesis is esterified by ACAT-2 for storage. In addition to suppression of cholesterol biosynthesis, the entry of cholesterol via the LDL pathway leads to downregulation of LDL receptors, an effect also observed with dietary saturated fat. Further regulatory mechanisms involve shortening of the residence time of LDL receptors in cell membranes The PCSK9 protein increases the proteolytic degradation of LDL receptors, decreasing the ability of cells to endocytose LDL. Another mechanism that leads to receptor degradation involves ubiquitination of the receptors by Idol (inducible degrader of the LDL receptor).
When isolated by ultracentrifugation, HDL appear to comprise two major classes: HDL2 and HDL3. Similar quantities of HDL3 are isolated from serum of men and women, but about twice as much HDL2 is found in premenopausal women. Immunochemical studies indicate that there are as many as 20 discrete species of HDL that are obscured by ultracentrifugation. One of these, the 67-kDa prebeta-1 HDL, is the primary acquisitor of cholesterol in the retrieval pathway from peripheral tissues.
Both liver and intestine produce HDL apolipoproteins, which organize with lipids into the native species of HDL in lymph and plasma. Excess free cholesterol and phospholipids liberated from the surface monolayers of chylomicrons and VLDL as hydrolysis of triglycerides proceeds are transferred to HDL by phospholipid transfer protein. Free cholesterol acquired by HDL is esterified by LCAT. This enzyme transfers 1 mol of fatty acid from a lecithin molecule to the hydroxyl group of unesterified cholesterol, forming cholesteryl esters. LCAT is secreted by liver. In severe hepatic parenchymal disease, levels in plasma are low and esterification of cholesterol is impeded, leading to the accumulation of free cholesterol in lipoproteins and in membranes of erythrocytes. This transforms them into the target cells classically associated with hepatic disease.
HDL serve as carriers for the C apolipoproteins, transferring them to nascent VLDL and chylomicrons. HDL as well as LDL deliver cholesterol to the adrenal cortex and gonads in support of steroidogenesis. HDL play a major role in the centripetal transport of cholesterol. Unesterified cholesterol, exported by the ATP-binding cassette transporter A1 (ABCA1), is acquired from the membranes of peripheral tissues by prebeta-1 HDL and is esterified by LCAT, passing through other HDL species before the cholesteryl esters are incorporated into HDL of alpha-electrophoretic mobility. Cholesterol effluxed from macrophages by the ATP-binding cassette transporter G1 (ABCG1) is acquired by larger HDL. The cholesteryl esters are then transferred to LDL and to triglyceride-rich lipoproteins mediated by cholesteryl ester transfer protein (CETP). Remnants of chylomicrons and a significant fraction of VLDL remnants and LDL are taken up by liver. Cholesteryl esters are also transferred from HDL to hepatocytes by scavenger receptor, class B, type I (SR-BI) receptors.
The pathways of catabolism of HDL are not yet known. Radiochemical studies indicate that Apo A-I and Apo A-II are removed from plasma synchronously and that a portion of the degradation occurs in liver and in kidney.
Cholesterol is an essential constituent of the plasma membranes of cells and of myelin. It is required for adrenal and gonadal steroidogenesis and for production of bile acids by liver. Cells synthesize cholesterol, commencing with acetyl-CoA. Formation of HMG-CoA is the initial step. The first committed step, mediated by HMG-CoA reductase, is the formation of mevalonic acid, which is then metabolized via a series of isoprenoid intermediaries to squalene. The latter cyclizes to form a series of sterols leading to cholesterol. A small amount of the mevalonate is converted to the isoprenoid substances ubiquinone, dolichol, and isopentenyl pyrophosphate. This pathway also yields the isoprenoid intermediaries geranyl and farnesyl pyrophosphates that are involved in prenylation of proteins. Prenylation provides an anchor so that proteins such as the low molecular weight G proteins can bind to membranes. Cholesterol synthesis is tightly regulated by cholesterol or its metabolites, which down regulate HMG-CoA reductase. Thus, cells can produce cholesterol not provided by circulating lipoproteins. Hepatocytes and intestinal epithelial cells use cholesterol for secretion of lipoproteins. In addition, cells constantly transfer cholesterol to circulating lipoproteins, chiefly HDL. Cholesterol is converted to bile acids in liver via a pathway initiated by cholesterol 7α-hydroxylase. Most of the bile acids are reabsorbed from the intestine, but the small amount that is lost in stool provides a means of elimination of cholesterol. Activity of cholesterol 7α-hydroxylase is decreased in hypothyroidism, as are the expression of LDL receptors and hepatic lipase.
At usual levels of cholesterol intake, about one-third of the amount ingested is absorbed. Most is transported to liver in chylomicron remnants, suppressing hepatic cholesterogenesis. Individuals may differ substantially in the effect of dietary cholesterol on levels of serum lipoproteins.
Because chylomicrons normally may be present in plasma up to 10 hours after a meal, they contribute as much as 600 mg/dL (6.9 mmol/L) to the triglycerides measured during that period. This alimentary lipemia can be prolonged if alcohol is consumed with the meal. Thus, serum lipids and lipoproteins should be measured after a 10-hour fast. If blood glucose is not to be measured, patients may have fruit and black coffee with sugar (which provide no triglyceride) for breakfast.
Much useful information is gained from inspection of the serum, especially before and after overnight refrigeration. Opalescence is due to light scattering by large triglyceride-rich lipoproteins. Serum begins to appear hazy when the level of triglycerides reaches 200 mg/dL (2.3 mmol/L). Chylomicrons are readily detected, because they form a white supernatant layer after refrigeration. Uncommon cases in which binding of immunoglobulins to lipoproteins takes place can be detected by the formation of a curd-like lipoprotein aggregate or a snowy precipitate as serum cools. If one of these disorders is suspected, blood should be kept at 37°C during the formation of the clot and separation of the serum, because the critical temperatures for precipitation of the cryoglobulin complex may be higher than room temperature.
Several chemical techniques provide reliable measures of cholesterol and triglycerides, an essential minimum for differentiation of disorders of lipoproteins. Unesterified and esterified cholesterol are usually measured together, so that the reported value is the total content of cholesterol in serum. A more complete characterization of lipoproteins is achieved by measurement of the cholesterol and triglyceride contents of individual lipoprotein fractions, separated by preparative ultracentrifugation, a technique usually available only in research laboratories. An efficient quantitative method employs vertical rotor ultracentrifugation, affording assessment of lipoprotein concentrations and particle diameters. LDL particle size can also be assessed by electrophoresis. The content of HDL can be measured using a technique in which they are the only lipoproteins that remain in solution after treatment of the serum with heparin and manganese. Although rapid, the results of this technique tend to be unacceptably variable unless rigid quality control is exercised. Prognostic implications of small changes in HDL cholesterol make such controls necessary. An important determinant of the content of cholesteryl esters in HDL is the amount of triglyceride-rich lipoproteins to which the HDL are exposed in plasma. Cholesteryl esters from HDL transfer into triglyceride-rich lipoproteins, leading to an inverse dependence of HDL cholesterol on plasma triglycerides. HDL cholesterol cannot be interpreted without knowledge of the level of serum triglycerides. For example, a level of HDL that would normally contain 45 mg/dL (1.17 mmol/L) of cholesterol might contain 37 mg/dL (0.96 mmol/L) when the triglycerides were 200 mg/dL (2.3 mmol/L) and 30 mg/dL (0.78 mmol/L) when they reach 500 mg/dL (5.7 mmol/L).
More sophisticated tests of composition of isolated lipoprotein fractions are of use in certain instances, including the ratio of cholesterol to triglycerides. Enrichment of VLDL by cholesteryl esters is typical of familial dysbetalipoproteinemia that is due to homozygosity for Apo E-2. The Apo E genotype can be determined by PCR analysis. Clinically useful immunoassays are available for Apo B and Lp(a).
Epidemiologic evidence suggests that LDL particles of smaller than normal diameter are associated with an increased risk of atherosclerosis. Small, dense LDL particles are a constant finding when triglyceride levels are elevated even marginally. Laboratory measurement of LDL diameters is therefore unnecessary in patients with hypertriglyceridemia.
Serum cholesterol and triglyceride levels are continuously distributed in the population; therefore, some arbitrary levels must be established to define significance. Epidemiologic studies in Europe and the United States have shown a progressive increase in risk of coronary disease as levels of cholesterol increase. Physicians should at least encourage patients at risk to eat diets low in saturated fats, trans fats, and cholesterol to minimize the burden of LDL in plasma.
The National Cholesterol Education Program has developed guidelines for treatment of hypercholesterolemia in adults. Since this 2001 report, the results of major clinical trials have suggested that more aggressive lipid management is justified. For patients at high risk (coronary heart disease [CHD] or CHD risk equivalents, including diabetes), a suggested LDL-cholesterol goal is less than 70 mg/dL (1.8 mmol/L). Triglyceride levels above 150 mg/dL (1.7 mmol/L) merit investigation. One abnormality commonly associated with increased risk of coronary disease that is not detected if screening is limited to hyperlipidemia is hypoalphalipoproteinemia, or deficiency of HDL. Many affected individuals have normal levels of cholesterol and triglycerides and no clinical features to alert the physician. HDL deficiency underscores the importance of controlling other risk factors and avoiding factors that reduce HDL, such as smoking, the use of some drugs, and obesity.
The second step in investigation of hyperlipidemia is determination of the species of lipoproteins involved. In some cases, this may include multiple species; in others, qualitative properties of the lipoproteins are of diagnostic importance. Secondary hyperlipidemias of similar pattern may be the sole cause of the lipoprotein abnormality or may aggravate primary disorders of lipoprotein metabolism. The differentiation of specific primary disorders usually requires additional clinical and genetic information.
The following diagnostic protocol, based on initial measurement of cholesterol, triglycerides, and HDL in serum after a 10-hour fast, supplemented by observation of serum and by additional laboratory measurements where essential, serves as a guide in identifying abnormal patterns. The term hyperlipidemia denotes high levels of any class of lipoprotein; hyperlipemia denotes high levels of any of the triglyceride-rich lipoproteins.
If the serum cholesterol level is modestly elevated (up to 260 mg/dL [6.72 mmol/L]), elevated levels of HDL may account for the observed increase in serum cholesterol. This is usually not associated with disease processes. The LDL cholesterol (in mg/dL) may be estimated by subtracting the HDL cholesterol and the estimated cholesterol contribution of VLDL from the total cholesterol level. The VLDL cholesterol is approximated as one-fifth of the serum triglyceride level.
Very high levels of HDL measured by the precipitation technique can signal the presence of the abnormal lipoprotein of cholestasis (Lp-X). This disorder is characterized by elevated alkaline phosphatase activity. Rarely, deficiency of CETP or hepatic lipase can cause high levels of HDL.
Here it is apparent that the primary abnormality is an increase in triglyceride-rich VLDL (hyperprebetalipoproteinemia) or chylomicrons (chylomicronemia), or both (mixed lipemia). Because both contain free cholesterol in their surface monolayers and a small amount of cholesteryl ester in their cores, the total cholesterol may be increased, although to a much smaller extent than triglycerides. Low levels of LDL often seen in hypertriglyceridemia may offset the increase in cholesterol due to the triglyceride-rich lipoproteins, especially in primary chylomicronemia. Because VLDL and chylomicrons compete as substrates in a common removal pathway, chylomicrons are nearly always present when triglyceride levels exceed 1000 mg/dL (11.5 mmol/L).
This pattern can be the result of either of two abnormal phenotypes. One is a combined increase of VLDL and LDL. This pattern is termed combined hyperlipidemia and is one of the three phenotypic patterns encountered in kindreds with the disorder FCH. The second phenotype is an increase of remnant lipoproteins derived from VLDL and chylomicrons. These particles have been partially depleted of triglyceride by LPL and enriched with cholesteryl esters by the LCAT system, such that the total content of cholesterol in serum is similar to that of triglycerides. This pattern is almost always an expression of familial dysbetalipoproteinemia. Diagnosis of this disorder is confirmed by a genotype demonstrating absence of the normal E-3 and E-4 alleles.
Clinical Descriptions of Primary and Secondary Disorders of Lipoprotein Metabolism
Epidemiologic evidence supports the atherogenicity of VLDL and their remnants. They have been demonstrated in atherosclerotic plaques from humans. Impaired capacity of the VLDL of some individuals to accept cholesteryl esters from the LCAT reaction may also contribute to atherogenesis by impeding centripetal transport of cholesterol.
Very high levels of triglycerides in plasma are associated with a risk of acute pancreatitis, probably from the local release of FFA and lysolecithin from lipoprotein substrates in the pancreatic capillary bed. When the concentrations of these lipids exceed the binding capacity of albumin, they could lyse membranes of parenchymal cells, initiating a chemical pancreatitis. Many patients with lipemia have intermittent episodes of epigastric pain during which serum amylase does not reach levels commonly considered diagnostic for pancreatitis. This is especially true in patients who have had previous attacks. The observation that these episodes frequently evolve into classic pancreatitis suggests that they represent incipient pancreatic inflammation. The progression of pancreatitis can be prevented by rapid reduction of triglycerides, usually accomplished by restriction of all dietary fat. In some cases, parenteral feeding, excluding fat emulsions, may be required for a few days. The clinical course of pancreatitis in patients with lipemia is typical of the general experience with this disease.
When triglyceride levels in serum exceed 3000 to 4000 mg/dL (34.5-46 mmol/L), light scattering by these particles in the blood lends a whitish cast to the venous vascular bed of the retina, a sign known as lipemia retinalis. Markedly elevated levels of VLDL or chylomicrons may be associated with the appearance of eruptive cutaneous xanthomas (see Figure 19–3E). These lesions, filled with foam cells, appear as yellow morbilliform eruptions 2 to 5 mm in diameter, often with erythematous areolae. They usually occur in clusters on extensor surfaces such as the elbows, knees, and buttocks. They are transient and disappear within a few weeks after triglyceride levels are reduced.
Figure 19–3
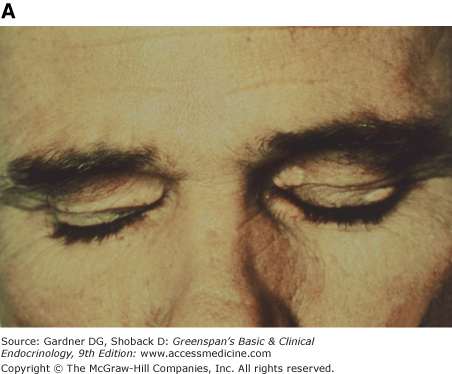
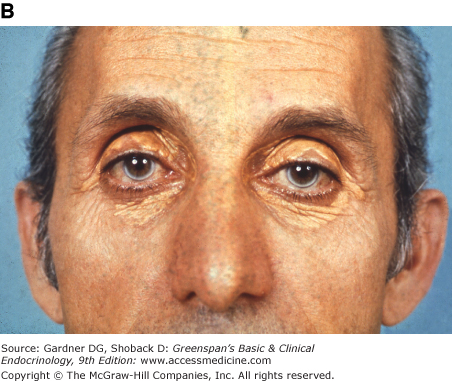
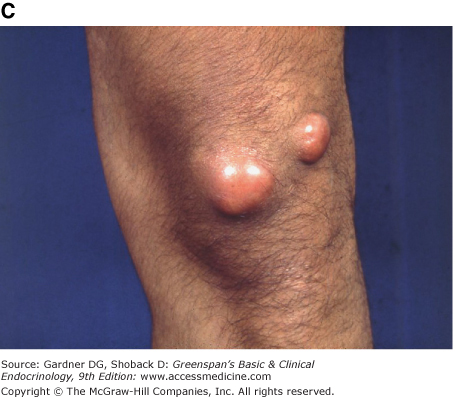
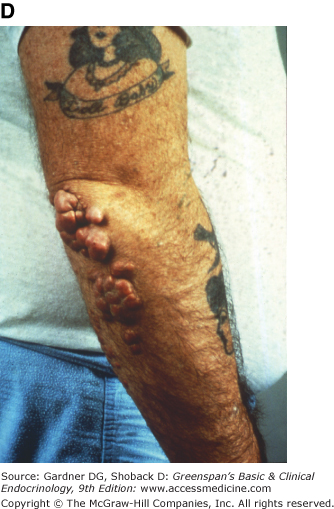
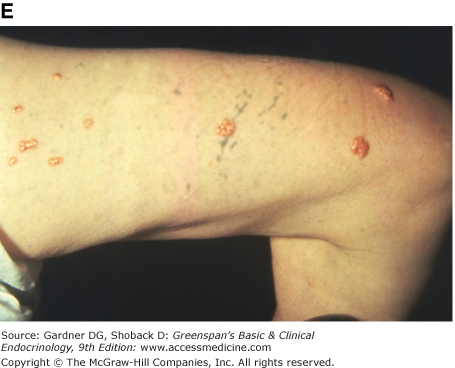
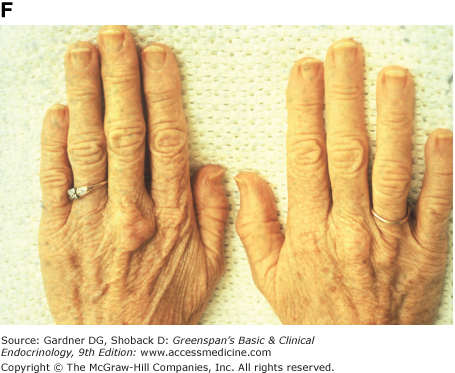
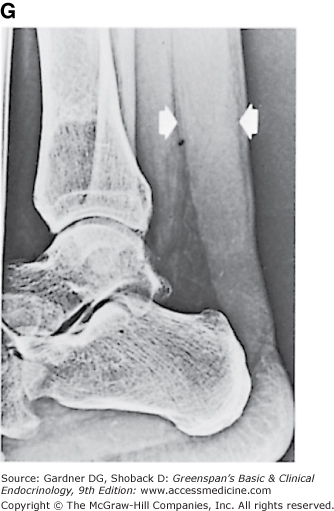
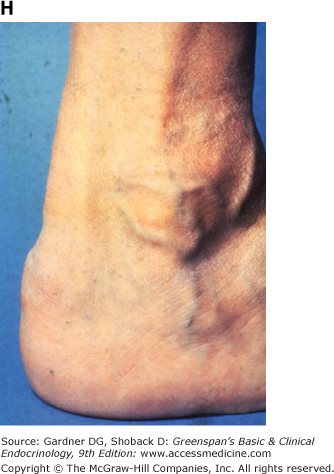
Clinical manifestations of hyperlipidemias. A. Xanthelasma involving medial and lateral canthi. B. Severe xanthelasma and arcus corneae. C. Tuberous xanthomas. D. Large tuberous xanthoma of elbow. E. Eruptive xanthomas, singly and in rosettes. F. Xanthomas of extensor tendons of the hands. G. Xeroradiogram of Achilles tendon xanthoma. H. Xanthoma of Achilles tendon. (Normal Achilles tendons do not exceed 7 mm in diameter in the region between the calcaneus and the point at which the tendon fibers begin to radiate toward their origins.)
Light scattering from high levels of triglyceride-rich lipoproteins can cause erroneous results in most chemical determinations involving photometric measurements. Amylase activity in serum may be inhibited; therefore, lipemic specimens should be diluted for measurement of this enzyme. Because the lipoproteins are not permeable to ionic or polar molecules, their core regions constitute a second phase in plasma. When the volume of this phase becomes appreciable, electrolytes and other hydrophilic species are underestimated with respect to their true concentration in the aqueous phase. A practical rule for correcting these values is as follows: For each 1000 mg/dL (11.5 mmol/L) of triglyceride, the measured concentrations of all hydrophilic molecules and ions should be adjusted upward by 1%.
The activity of LPL is dependent upon the integrity of the enzyme and its cofactor, Apo C-II. In addition, defects in Apo A-V, GPIHDLBP, and LMF1 proteins can result in severe impairment of LPL activity. The LMF1 protein is essential for the secretion of both LPL and hepatic lipase in their active conformations. Because the clinical expressions of these defects are similar, they are considered together. On a typical North American diet, lipemia is usually severe (triglycerides of 2000-25,000 mg/dL) (23-287.5 mmol/L). Hepatomegaly and splenomegaly are frequently present. Foam cells laden with lipid are found in liver, spleen, and bone marrow. Splenic infarct has been described and may be a source of abdominal pain. Hypersplenism with anemia, granulocytopenia, and thrombocytopenia can occur. Recurrent epigastric pain and overt pancreatitis are frequently encountered. Eruptive xanthomas may be present. These disorders may be recognized in early infancy or may go unnoticed until an attack of acute pancreatitis occurs or lipemic serum is noted on blood sampling as late as middle age. Patients with these disorders are usually not obese and have normal carbohydrate metabolism unless pancreatitis impairs insulinogenic capacity. Estrogens intensify the lipemia by stimulating hepatic production of VLDL. Therefore, in pregnancy and lactation or during the administration of estrogenic steroids, the risk of pancreatitis increases.
There is a preponderance of chylomicrons in serum such that the infranatant layer of serum refrigerated overnight may be nearly clear with a supernatant chylomicron layer. Many patients have a moderate increase in VLDL, however, and in pregnant women or those receiving estrogens, a pattern of mixed lipemia is usually present. Levels of LDL in serum are decreased, probably representing the predominant catabolism of VLDL by pathways that do not involve the production of LDL. Levels of HDL cholesterol are also decreased.
A presumptive diagnosis of these disorders can be made by restricting oral intake of fat to 10 to 15 g/d. Triglycerides drop precipitously, usually reaching 200 to 600 mg/dL (2.3-6.9 mmol/L) within 3 to 4 days. Confirmation of deficiency of LPL is obtained by measurement of the lipolytic activity of plasma 10 minutes after injection of heparin (0.2 mg/kg) intravenously. Analysis of lipolysis is done with and without 0.5 mol/L sodium chloride, which inhibits LPL but does not suppress the activity of hepatic lipase. Absence of the Apo C-II cofactor can be demonstrated most readily by electrophoresis or isoelectric focusing of the proteins of VLDL.
Treatment of primary chylomicronemia is entirely dietary. Intake of fat should be reduced to 10% or less of total calories. In an adult, this represents 15 to 30 g/d. Because the defect involves lipolysis, both saturated and unsaturated fats must be curtailed. The diet should contain at least 5 g of polyunsaturated fat as a source of essential fatty acids, and fat-soluble vitamins must be provided. Administration of 500 mg daily of marine omega-3 fatty acids is also recommended. Adherence to this diet invariably maintains triglycerides below 1000 mg/dL (11.2 mmol/L) in the absence of pregnancy, lactation, or the administration of exogenous estrogens. Because this is below the level at which pancreatitis usually occurs, compliant patients are at low risk. Pregnant women with these disorders require particularly close monitoring.
Endogenous lipemia (elevated VLDL) and mixed lipemia probably both result from several genetically determined disorders. Because VLDL and chylomicrons are competing substrates in the intravascular lipolytic pathway, saturating levels of VLDL impede the removal of chylomicrons. Therefore, as the severity of endogenous lipemia increases, a pattern of mixed lipemia may supervene. In other cases, the pattern of mixed lipemia appears to be present continuously. Although specific pathophysiologic mechanisms remain obscure, certain familial patterns are known. In all forms, factors that increase the rate of secretion of VLDL aggravate the hypertriglyceridemia (ie, obesity with insulin resistance, appearance of fully developed type 2 diabetes mellitus, alcohol, and exogenous estrogens). Studies of VLDL turnover indicate that either increased production or impaired removal of VLDL may be operative in different individuals. A substantial number of patients with mixed lipemia have partial defects in catabolism of triglyceride-rich lipoproteins, often due to heterozygosity for mutations in LPL. Most patients with significant endogenous or mixed lipemia have centripetal obesity.
Clinical features of these forms of hypertriglyceridemia depend on their severity and include eruptive xanthomas, lipemia retinalis, recurrent epigastric pain, and acute pancreatitis.
Endogenous lipemia is frequently an element in a constellation of metabolic abnormalities termed the metabolic syndrome. Insulin resistance, usually associated with central obesity, is a core feature of this syndrome. Patients may or may not have hyperglycemia. Low levels of HDL cholesterol largely reflect increased transfer of CE into circulating VLDL. Hypertension and hyperuricemia are frequently present.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


