Importance of the Intestine in Modulating the Cholesterol Content of Liver and LDL Metabolism
Chylomicrons are apoB-containing lipoproteins made in the intestine and contain apoB-48 as their major apolipoprotein (see also
Chapters 1 and
7). Lipids in the intestinal lumen such as TG, CE, FC, and PL are emulsified by bile salts and hydrolyzed by pancreatic lipases. Free fatty acids (FFA), monoacylglycerols, FC, and derivatives of PL are taken up by the intestinal cells, wherein TG are reformed by diacylglycerol transferase and CE by an acyl cholesterol acyltransferase (ACAT), respectively (see also
Chapter 7). The resultant TG and CE become associated with apoB-48 through the action of microsomal triglyceride transfer protein (MTP) (see also
Chapter 7).
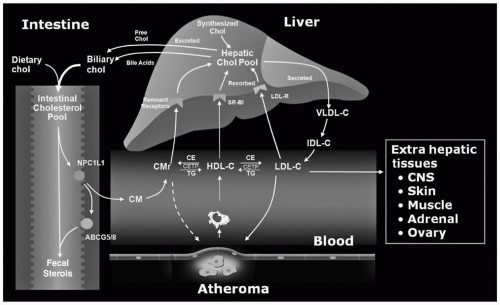
Cholesterol in the lumen of the intestine is derived from dietary (about 400 mg/day) and hepatic (about 1,100 mg/day in bile) origins. About half this sterol (range 29% to 80%) is absorbed by intestinal cells through the action of Niemann-Pick C1 Like 1 (NPC1L1) on the surface of enterocytes (
3), with the remaining half being excreted into the stool (
Fig. 8.1). Nonsynonymous sequence variants in NPC1L-1 contribute to significant variability in cholesterol absorption and plasma levels of low-density lipoprotein cholesterol (LDL-C). Excess absorption of sterol, either cholesterol or plant sterols, is prevented through the coordinate action of two half-transporters, ATP-binding cassette (ABC) proteins, ABCG5 and ABCG8, which pump sterols from inside the intestinal cells back into the lumen (
4) (
Fig. 8.1). Normal humans absorb <5% of dietary plant sterols (
4).
In contrast to cholesterol, most of the dietary TG is absorbed. The FFA derived from the TG may be saturated,
monounsaturated, polyunsaturated, or trans fatty acids. The nature of the fatty acid that is delivered to the liver can ultimately influence the hepatic cholesterol pool and LDL metabolism (see the following text).
ApoB-48 is necessary for secretion of chylomicrons from the intestinal cell into the lymphatics, from which they enter the systemic circulation through the thoracic duct. In the systemic circulation, TG in the core of the chylomicrons are rapidly hydrolyzed on the surface of the endothelial cells by lipoprotein lipase (LPL), which is attached to the cell membrane by heparin sulfate. Apolipoprotein C-II (apoC-II) is a cofactor for LPL and together they convert chylomicrons into chylomicron remnants (see also
Chapter 7). These remnants are smaller particles that still contain the cholesterol from the intestine but much less TG.
The chylomicron remnants are quickly removed from the circulation by the interaction of apolipoprotein E (apoE) with the chylomicron remnant receptor (also called the low-density lipoprotein-related receptor [LRP]) on the surface of the hepatocytes (
5) (
Fig. 8.1). The LDLR (see also the following text) and heparan-sulfate proteoglycans can also mediate the uptake of chylomicron remnants (
5). As cholesterol on the chylomicron remnants is taken up, the hepatic cholesterol pool increases, which then normally downregulates both the LDLR and endogenous cholesterol production (
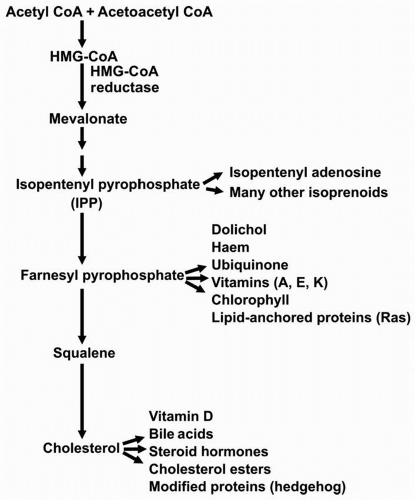
2). The rate-limiting enzyme of cholesterol biosynthesis is hydroxymethylglutaryl CoA (HMG-CoA) reductase that catalyzes the conversion of HMG-CoA to mevalonate (
2,
6) (
Fig. 8.2). A decrease in the number of LDLR leads to a decreased uptake of LDL and other TG-enriched apoB-containing particles. The precise pathway by which cell cholesterol regulates these processes through a transcription factor, the sterol regulatory element binding protein (SREBP) (
2), is discussed later in the text.
As TG are transported on chylomicrons and hydrolyzed, FFA are produced and taken up either by adipose tissue, where they are re-esterified into TG and stored for future energy use, or by skeletal muscle for immediate energy use. However, some TG are still associated with the chylomicron remnant. As the remnant particle is taken up by the liver, the TG are hydrolyzed, producing FFA. Monounsaturated fatty acids are the preferred substrate for ACAT, the enzyme that esterifies cholesterol, thereby decreasing the hepatic pool of cholesterol and consequently upregulating LDLR and lowering LDL-C
levels (
1). In contrast, saturated fatty acids such as palmitic acid appear to interfere with LDLR-mediated clearance of LDL-C and raise LDL-C (
1). The precise mechanism of this action of saturated fats is not completely elucidated. Hypotheses include: (i) decreased esterification of saturated fatty acids to cholesterol by ACAT, thereby increasing the FC pool, decreasing LDLR, and increasing LDL-C; (ii) enrichment of cell membrane PL with saturated fats, interfering with the normal function of LDLR; and (iii) redistribution of FC by saturated fats into a compartment that favors the inhibitory effect of FC on LDLR (
1).
Thus, both the cholesterol content and the nature of the FFA in the TG in chylomicrons can influence the cholesterol pool in liver and the number of LDLR.
Importance of the Liver in Modulating its Cholesterol Content and LDL Metabolism
The liver can synthesize cholesterol from acetyl CoA, which can be derived from fat, protein, or carbohydrate metabolism, and used for the production of cholesterol and a number of other molecules derived from mevalonic acid (
Fig. 8.2). As the cholesterol pool in the liver increases, expression of HMG-CoA reductase is downregulated and cholesterol synthesis decreases. Conversely, as the hepatic cholesterol pool decreases, expression of HMG-CoA reductase is upregulated and cholesterol biosynthesis increases. The gene for the LDLR is similarly regulated by the cholesterol content of liver. In fact, transcription of the genes for HMG-CoA reductase and the LDLR is coordinately regulated since each gene promoter has a sterol response element (SRE) that responds in a similar fashion to SREBP (
2).
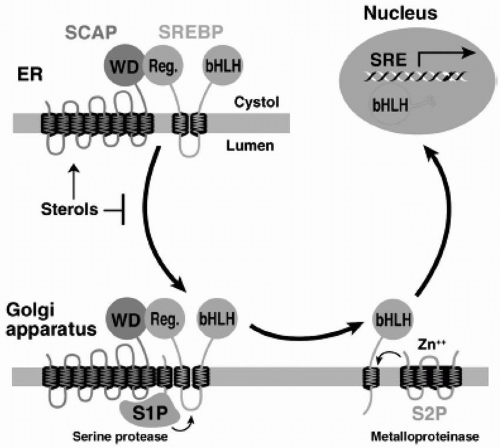
The SREBP Pathway. SREBP is an endoplasmic reticulum (ER) membrane-bound transcription factor that controls expression of genes required for cholesterol synthesis and uptake (
7). The amount of FC in the liver regulates the proteolytic release of active SREBP from the membrane (
2) (
Fig. 8.3). This cholesterol regulation occurs through the SREBP cleavage activating protein (SCAP) that is both a sensor of sterols and an intracellular escort of SREBP. When hepatocytes are depleted of cholesterol, SCAP transports SREBP from the ER to the Golgi, where two proteases, Site-1 protease (S1P) and Site-2 protease (S2P), act in sequence to release the NH2-terminal transcription factor domain of SREBP from the membrane (
2) (
Fig. 8.3). The NH2-terminus of SREBP containing the bHLH-Zip domain of SREBP enters the nucleus and binds to an SRE in the promoter of the LDLR and HMG-CoA reductase genes, increasing their transcription. As the cholesterol content of the hepatocyte increases, the SREBP/SCAP complex is not incorporated into ER transport vesicles, SREBP cannot reach the Golgi, the NH2-terminal domain of SREBP cannot be released from the membrane for transport into the nucleus, and the transcription of the LDLR and HMG-CoA reductase genes decreases (
2).
The liver is sensitive to the uptake of cholesterol from any particular pathway. This includes the low density lipoprotein receptor-related protein (LRP) (also called the chylomicron remnant receptor) (see
Fig. 8.1 and preceding text), the LDLR, and Scavenger receptor class B type I (SR-B1), which promotes the uptake of CE and FC from high-density lipoprotein (HDL) (see the following text).
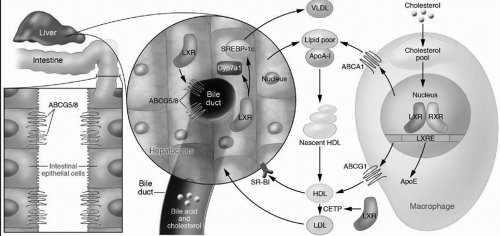
Liver X receptor (LXR). In addition to downregulating HMG-CoA reductase and LDLR, the liver can decrease its cholesterol content by exporting cholesterol directly into bile, by converting cholesterol into bile acids, by upregulating cell surface proteins such as ABCA1 (see also the following text) to increase the removal of intracellular cholesterol onto lipoprotein acceptors, and by utilizing cell cholesterol for newly synthesized very-low-density lipoproteins (VLDL). The LXR, a nuclear receptor, stimulates all of these processes (
8) (
Fig. 8.4). It is activated by sensing oxysterols that are produced as cellular cholesterol increases. Activated LXR increases the expression of the ABCG5/ABCG8 transporters (see also the preceding text), which then excrete cholesterol directly into the bile. In mice but not in humans LXR also stimulates the expression of 7α-hydroxylase, the rate-limiting enzyme of bile acid synthesis, leading to increased conversion of cholesterol into bile acids for excretion into bile. The upregulation of ABCA1 by LXR is also important in macrophages to promote the egress of excess cholesterol onto lipid-poor apoA-I (
8) (
Fig. 8.4) (see also the following text). Finally, LXR also stimulates VLDL production (
Fig. 8.4) by the upregulation of the master regulator of hepatic lipogenesis, SREBP-1c. that turns on the transcription of genes utilized for fatty acid synthesis, and subsequently, for TG and PL biosynthesis (
9).
Farnesoid X Receptor (FXR). The nuclear receptor FXR is activated by binding to bile acids in the cells of either the intestine or the liver and plays a paramount role in the enterohepatic circulation of bile acids (
8). Activated FXR increases the
expression of bile acid-binding proteins and transports proteins that play a role in the import and export of bile acids in cells. FXR controls bile acid synthesis by suppressing cholesterol 7α-hydroxylase expression, thereby decreasing the conversion of cholesterol into bile acids (
8) (
Fig. 8.4).
Biosynthesis and Secretion of VLDL. The biosynthesis of VLDL requires the production of TG, CE, and apoB-100, and the action of medium chain triglycerides (MCT). The production of TG is often increased when there is an increased flux of FFA from adipose tissue to liver. This normally occurs in the fasting state when insulin levels are lower and the activity of hormone-sensitive lipase (HSL) in adipocytes is increased. Lower insulin levels also lead to increased biosynthesis of cholesterol and CE. MCT permits the interaction of these lipid moieties with apoB-100, thereby decreasing its proteolytic breakdown (apoB-100 is constitutively expressed). This leads to increased VLDL production and secretion into blood.
Once in the systemic circulation, the TG on VLDL undergo the same hydrolytic process as do chylomicrons (see the preceding text) (
Fig. 8.1). VLDL remnants are formed. The smaller VLDL remnant, intermediate-density lipoproteins (IDL), can be taken up by the LDLR on the surface of liver or converted by hepatic lipase (HL) into LDL itself (which is then also taken up by LDLR on liver or peripheral cells) (
Fig. 8.1). The VLDL-IDL-LDL pathway is important to regulate the cholesterol pool in the liver. Also, the biogenesis of LDL is derived from the catabolism of the TG-enriched VLDL. This will be critically important when we examine the disorders of LDL overproduction.
Role of the ApoA-I-containing Lipoproteins in LDL Metabolism
Most of the plasma apoA-I is associated with HDL (
10) (see also
Chapter 9). In fact, apoA-I comprises about 70% to 80% of the apolipoprotein content of HDL. ApoA-I is synthesized in both the liver and intestine where it is secreted into blood as a very lipid-poor molecule (
8,
10) (
Fig. 8.4) (see also
Chapter 9). ApoA-I interacts with ABCA1 on the surface of peripheral cells, such as macrophages, catalyzing the removal of FC and PL across the cell membrane and forming a nascent HDL particle consisting of apoA-I, FC, and PL (
Fig. 8.4). As more lipid is accrued from cell membranes and from the transfer of lipid from TG-rich lipoproteins by the phospholipid transfer protein (PLTP), the activity of lecithin-cholesterol acyl transferase (LCAT) increases (see
Chapter 9). LCAT with its cofactor apoA-I removes a fatty acid from lecithin and esterifies it to FC, producing CE and lysolecithin. As the amount of CE in the core of the HDL particle increases, its
shape changes from a cigar-shaped nascent HDL particle to a mature, spherical HDL (
Fig. 8.4).
Transfer of CE from the Core of HDL to the Core of ApoB-containing Lipoproteins, Including LDL. As the CE in the core of the mature HDL increases, some CE is transferred from HDL to the apoB-containing lipoproteins in exchange for TG via the action of the cholesteryl ester transfer protein (CETP) (
Fig. 8.1) (
10). The CE is then taken up by the liver through the LDLR, increasing the hepatic cholesterol pool. In humans, over half the cholesterol removed from peripheral cells by apoA-I is returned to the liver by the apoB-containing lipoproteins via the LDLR (
Fig. 8.1). The remainder of the CE on HDL is delivered to the liver directly through the interaction of apoA-I with the SR-BI receptor that catalyzes the “dumping” of CE and FC into liver. HDL is then released back into the circulation. HDL can also remove cholesterol from cells via ABCG1 (
Fig. 8.4) (
10) (see also
Chapter 9).
The entire process of returning cholesterol from peripheral cells to the liver for excretion into bile is termed the “reverse cholesterol transport” (RCT) pathway (
10). Once the cholesterol is delivered to the liver, it must then be excreted from the liver into the bile or converted to bile acids to complete RCT (
10). Cholesterol can be delivered to the liver via SR-BI receptor or by its uptake as part of the internalization of apoB-containing lipoproteins by the LDLR. Thus, the LDLR pathway (see also the following text) plays an important role in the RCT process and the metabolism of the apoA-I-containing lipoproteins (
Figs. 8.1 and
8.4).
Discovery of the LDLR
In the 1970s, Michael Brown and Joseph Goldstein performed a series of elegant experiments that led to the discovery of the LDLR (
2) and to their receipt of the Nobel Prize in Medicine or Physiology in 1985. Even then, the paramount importance of the liver in cholesterol metabolism was appreciated. For example, cholesterol synthesis was known to be controlled by amount of cholesterol in diet through an end-product, negative feedback mechanism. The rate-limiting step in cholesterol synthesis was known to be the synthesis of mevalonic acid by HMG-CoA reductase. However, Drs. Goldstein and Brown developed the human fibroblast as a cell culture model to study cholesterol metabolism and established an assay to measure HMG-CoA reductase activity in fibroblasts. In addition, they selected fibroblasts from patients with the rare homozygous form of familial hypercholesterolemia (FH) and their obligate heterozygous FH parents for study. FH homozygotes had profound elevations in LDL-C of 600 to 900 mg/dL, while lesser but still impressive increases in LDL-C of 300 to 400 mg/dL were present in FH heterozygotes (see also the following text).
A very low rate of HMG-CoA reductase activity was found in normal fibroblasts in the presence of fetal calf serum (that contained cholesterol on lipoproteins). When the serum was removed from the medium, there was over a 50-fold increase in the specific activity of HMG-CoA reductase within 16 hours (
2). When normal and FH homozygous fibroblasts were compared in the presence of fetal calf serum, there was 75-fold greater HMG-CoA reductase activity in the FH cells than in normal cells. When the cell culture medium containing fetal
calf serum was subsequently switched to lipoprotein-deficient serum, HMG-CoA reductase activity increased markedly in the normal cells, while in the FH homozygous cells the activity remained elevated. When LDL was subsequently added to the medium, there was a rapid inhibition of HMG-CoA reductase activity in normal cells, but no change in the mutant FH cells. Thus, the FH homozygous cells had a defect in the regulation of HMG-CoA reductase. The rest is history (
2,
11). The overproduction of cholesterol in FH patients was not due to a defect in HMG-CoA reductase, but rather to a defect in the binding and uptake of LDL, such that LDL could not enter the cells and inhibit cholesterol biosynthesis.
LDLR Pathway
Over the next decade, Brown and Goldstein systematically worked out the LDLR pathway (
2,
11) (see
Fig. 1.3, p. 7) and elucidated the structure of the LDLR. The LDLR is synthesized as a 120-kDa precursor. After glycosylation in the ER and Golgi, the mature 140-kDa LDLR reaches the cell surface where it is directed toward the clathrin-coated pits (
Fig. 1.3). The crystal structure of the extracellular domain of the LDLR has provided insight into how the LDLR binds LDL with high affinity at the cell membrane and then releases LDL in the appropriate intracellular compartment (
2,
6,
11). The extracellular domain consists of a ligand-binding domain, an epidermal growth factor (EGF) precursor homology domain (that contains a six-bladed β-propeller flanked by cysteine-rich EGF repeats), and an O-linked sugar-rich domain. When the LDLR is in clathrin pits on the cell surface, its extracellular domain is extended, exposing the ligand-binding domain of the LDLR to LDL. After the LDLR binds the apoB-100 of LDL, the receptor-ligand complex is internalized within coated vesicles by endocytosis and delivered to endosomes. An adaptor protein called autosomal recessive hypercholesterolemia (ARH) after the disorder that permitted its discovery (see also the following text) and also known as the LDLR adaptor protein couples the LDLR to the endocytic machinery, permitting LDLR internalization (
12). In the acidic environment of the endosome, the LDLR folds back on itself, and the β-propeller region of the epidermal growth factor (EGF) precursor domain is brought into close apposition to the ligand-binding domain, thereby displacing LDL and permitting release of LDL in the endosome and recycling of the LDLR to the cell surface for reuse (
6,
10,
11). The released LDL is subsequently degraded in the lysosome (
Figs. 1.3 and
1.5). The apoB-100 moiety undergoes proteolysis and CE are hydrolyzed into FC and FFA. The FC derived from LDL decreases HMG-CoA reductase and LDLR activity by inhibiting the SREBP pathway (see the preceding text), which was subsequently elucidated in the 1980s and 1990s (
2,
6,
11).
In addition to the apoB-100 moiety on LDL, the LDLR also binds with high affinity to apoE on TG-rich lipoproteins, promoting their internalization and cellular metabolism. This process appears to occur independently of ARH (
12).
Within the paradigm of the LDLR pathway, it was shown that there were five functional classes of mutations in the
LDLR, namely those that affected either the synthesis, intracellular transport, binding, internalization, or recycling of the LDLR. Over 900 mutations have been described in the
LDLR (
12) (see also the following text).
Proprotein Convertase Subtilisin-Like Kexin Type 9 Pathway
Proprotein convertase subtilisin-like kexin type 9 (PCSK9) is the ninth member of secretory mammalian kexin-like subtilases that cleave a variety of precursor proteins. PCSK9 was initially described as a proteinase K-like subtilase, neural apoptosis-regulated convertase 1 (NARC-1) (
13). Patients with an autosomal dominant phenotype indistinguishable from FH (also called FH3) (see the following text) were found to have defects in PCSK9 (
14). Consistent with this finding, FH3 did not segregate with the LDLR or apoB genes (
Table 8.1). Insomuch as the study of FH patients led to the elucidation of the LDLR pathway (see the preceding text), experiments
with FH3 patients have led to a working hypothesis of how PCSK9 functions in LDL clearance.
Although compelling evidence indicates that PCSK9 negatively regulates the LDLR pathway, its precise role remains incompletely defined (
15). ProPCSK9 of 74 kDa undergoes intramolecular cleavage in the ER (
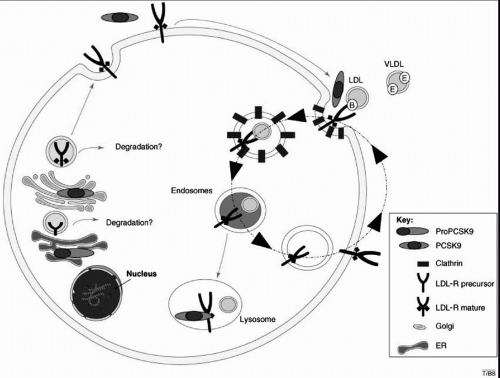
Fig. 8.5), producing a 14-kDa prodomain and a 60-kDa fragment. Both fragments remain bound by noncovalent links, and both are sulfated and glycosylated as they traverse the Golgi and exit the hepatocyte via the secretory pathway (
16). The crystal structure of PCSK9 indicates that this enzyme contains an active site catalytic triad, but that the N-terminal extension of the prodomain blocks access to the active site. Thus, it appears unlikely that PCSK9 functions as a protease to directly degrade the LDLR in either the ER or the Golgi (
Fig. 8.5).
PCSK9 binds specifically to the LDLR at its EGF-like repeat on the cell surface. Following endocytosis the PCSK9/LDLR complex is directed toward the lysosomes for degradation, which would decrease the recycling of the LDLR to the cell surface (
Figs. 1.3 and
1.5). PCSK9 has also been proposed to bind to LDLR within the secretory pathway and target it for lysosomal degradation. PCSK9 catalytic activity is not necessary for binding to the LDLR, indicating that PCSK9 does not degrade the LDLR at the cell surface but may act as a chaperone protein for the LDLR (
15,
16). Some gain-of-function mutations in PCSK9 increase PCSK9 affinity for the LDLR, while loss-of-function mutations have reduced affinity for the LDLR (see also the following text).
Regulation of PCSK9. The transcriptional regulation of PCSK9 is complex, with positive and negative regulation from insulin and different nuclear receptors (
16). Notably, like the LDLR, PCSK9 expression is downregulated by cholesterol and upregulated by statins via the SREBP pathway (
16). Thus,
while statin treatment increases LDLR expression, the effect of this increase is dampened by elevated PCSK9 expression. Consequently, the development of an inhibitor to PCSK9 action may significantly potentiate the effect of statins (
15).
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia and Atherosclerosis in Women
Dyslipidemia and Atherosclerosis in Women
 Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
Lipid, Apolipoprotein, and Lipoprotein Metabolism: Implications for the Diagnosis and Treatment of Dyslipidemia
 Disorders of Hypertriglyceridemia
Disorders of Hypertriglyceridemia
 Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
 Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease



