Abstract
This chapter describes the physiology of hemostasis including platelet plug formation, platelet vessel interaction, coagulation cascade and fibrinolysis; diagnosis and management of von Willebrand disease; inherited and rare bleeding disorders and hemophilia carrier detection; acquired clotting factor deficiencies; inhibitors associated with factors VIII and IX deficiencies and acquired inhibitors and their management; hemostasis in the newborn; mechanism of thrombosis in inherited thrombophilia, clinical manifestations, diagnostic testing and management of the hypercoagulable state, venous and arterial thrombosis and pulmonary embolism, hereditary thrombotic disorders, acquired disorders associated with increased risk of thrombosis, catheter-related thrombosis, lupus anticoagulant and the antiphospholipid syndrome and its management, thrombotic disorders and stroke in the newborn, antithrombotic, thrombolytic and antiplatelet agents, predisposition to thrombosis in general and prophylactic anticoagulation in high-risk situations.
Keywords
Hemostasis, von Willebrand disease, hemophilia, inhibitors, rare bleeding disorders, vitamin K deficiency, disseminated intravascular coagulation, thrombosis, stroke, hypercoagulable state, inherited thrombophilia
Hemostatic Disorders
Physiology of Hemostasis
As a result of injury to the blood vessel endothelium, three events take place concurrently:
- 1.
Vasoconstriction (vascular phase).
- 2.
Platelet plug formation (primary hemostatic mechanism—platelet phase).
- 3.
Fibrin thrombus formation (initiation, amplification, and propagation phases).
Relevant Components of Hemostasis
There are three integral components to hemostasis:
- 1.
Endothelial cells.
- 2.
Platelets.
- 3.
Plasma coagulation factors.
Endothelial cells secrete substances that:
- •
Repel platelets (prostaglandin I2 (PGI2), adenosine diphosphate (ADP), and nitric oxide).
- •
Initiate coagulation (collagen, fibronectin).
- •
Promote platelet adhesion (von Willebrand factor (vWF)) and fibrin dissolution (tissue plasminogen activator, t-PA).
- •
Catalyze the inhibition of thrombin (heparin and thrombomodulin).
- •
Inhibit the initiation of fibrin dissolution (t-PA inhibitor).
Participation of platelets in hemostasis is a fundamental component of the physiologic process of coagulation. Platelet interactions in coagulation are initiated by adhesion to areas of vascular injury. Subsequent activation of platelets results in release of ADP, serotonin, and calcium from “dense bodies” and fibrinogen, vWF, factor V (FV), high-molecular-weight (HMW) kininogen, fibronectin, α-1-antitrypsin, α-thromboglobulin, platelet factor 4 (PF4), and platelet-derived growth factor from α granules. Platelets provide surfaces for the assembly of coagulation factors (e.g. VIIIa/Ca 2+ /IXa and Va/Ca 2+ /Xa complexes). The platelets aggregate and increase the mass of the hemostatic plug. They also mediate blood vessel constriction (by releasing serotonin) and neutralize heparin.
All of the plasma coagulation factors are produced in the liver; factor VIII is also produced by endothelial cells. Table 15.1 lists the half-life and plasma levels of the coagulation factors. Factors II, VII, IX, and X are vitamin-K-dependent and require vitamin K in order to undergo post-translational gamma carboxylation. These vitamin-K-dependent factors circulate in zymogen form, are activated on platelet phospholipid surfaces and, upon activation, have serine protease activity. The plasma coagulation factors work in an interdependent manner to generate thrombin (factor IIa) from prothrombin (factor II); thrombin then converts fibrinogen to form fibrin monomers. Fibrin monomers polymerize and establish a network. Thrombin activates factor XIII which in turn crosslinks the fibrin network. By incorporating into the hemostatic plug, thrombin becomes inactivated. Thrombin plays a central bioregulatory role, promoting platelet aggregation and release reactions and generating a biofeedback-positive loop to form more thrombin at a faster rate. Thrombin and thrombin complexed to thrombomodulin also activate thrombin-activatable fibrinolysis inhibitor (TAFI), a procarboxypeptidase found in plasma, which attenuates fibrinolysis of the clot. The three components of hemostasis (blood vessels, platelets, and plasma coagulation factors) do not function independently but are integrated and interdependent.
| Factors | Common name | Biologic half-life (h) | Plasma concentration (nM) | Plasma levels (units/dl) |
|---|---|---|---|---|
| I | Fibrinogen | 56–82 | 8800 | 200–400 a |
| II | Prothrombin b | 45–60 | 1400 | 50–150 |
| III | Tissue thromboplastin | N/A | — | 0 |
| V | Proaccelerin, labile factor | 36 | 20 | 50–150 |
| VII | Proconvertin b , stable factor | 5 | 10 | 50–150 |
| VIII | Antihemophilic factor | 8–12 | 0.7 | 50–150 |
| IX | Christmas factor b | 12–24 | 90 | 50–150 |
| X | Stuart factor b | 24–60 | 170 | 50–150 |
| XI | Plasma thromboplastin antecedent | 48 | 30 | 50–150 |
| XII | Hageman factor | 48–52 | 375 | 50–150 |
| XIII | Fibrin-stabilizing factor | 168–240 | 70 | 50–150 |
| HMW kininogen | Fitzgerald factor | 136 | 6,000 | — |
| Prekallikrein | Flecher factor | N/A | 450 | — |
Primary Hemostatic Mechanism (Platelet Phase)
This mechanism leads to the formation of a reversible aggregate of platelets: a temporary hemostatic plug. Endothelial injury exposes vWF and collagen from the subendothelial matrix to flowing blood and shear forces. Plasma vWF binds to the exposed collagen, uncoils its structure and, in synergy with collagen supports the adhesion of platelets. Initially the vWF interacts with the GPIb platelet receptor, tethering the platelets. As the platelet collagen receptors GPVI and α2β1 bind to collagen, the platelets adhere and become activated with a resulting release of platelet alpha and dense granule contents. Platelet activation results in a conformational change in the αIIbβ3 receptor, activating it and enhancing its avidity for vWF, for vessel wall ligands and for fibrinogen. The enhanced avidity for vWF and fibrinogen mediates platelet-to-platelet interactions which eventually lead to platelet plug formation.
Fibrin Thrombus Formation
The fibrin thrombus formation component of hemostasis occurs in three overlapping phases ( Figure 15.1 ):
- 1.
Initiation.
- 2.
Amplification.
- 3.
Propagation.
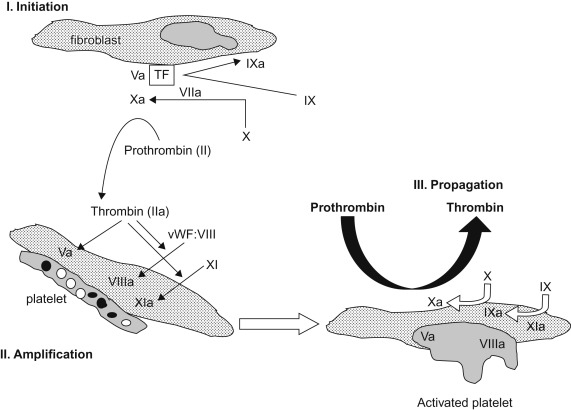
The initiation phase begins with cell-based expression of tissue factor (TF) at the site of endothelial injury. Factor VII binds to the exposed TF and is rapidly activated. The factor VIIa/TF complex in turn generates factor Xa (FXa) and factor IXa (FIXa). FXa can activate FV which complexes with FXa and generates small amounts of thrombin. During the amplification phase the procoagulant stimulus is transferred to the surface of platelets at the site of injury. The small amounts of thrombin enhance platelet adhesion, fully activate the platelets and activate factors V, VIII, and XI. In the propagation phase the “tenase” complex of FIXa–FVIIIa is assembled on the platelet surface and efficiently generates FXa. Similarly the “prothrombinase” complex of FXa–FVa is assembled on the platelet surface and efficiently generates thrombin. Unlike FXa generated from TF–FVIIa interactions, FXa complexed to FV is protected from inactivation by tissue factor pathway inhibitor (TFPI), assuring adequate thrombin generation. The resulting procoagulant, thrombin, activates factor XIII and cleaves fibrinopeptides (FPs) A and B from fibrinogen. The residual peptide chains aggregate by means of loose hydrogen bonds to form fibrin monomers. Under the influence of FXIIIa, fibrin monomers are converted into fibrin polymers, forming a stable fibrin clot. In the presence of thrombin, the mass of loosely aggregated intact platelets is transformed into a densely packed mass that is bound together by strands of fibrin to form a definitive hemostatic barrier against the loss of blood.
Platelet Vessel Interaction
The vasculature forms a circuit which maintains blood in a fluid state, free of leaks. With vascular injury, platelets and the coagulation system temporarily close the rent and repair the leak. Blood vessel wall characteristics exhibit properties that contribute to hemostasis or stop hemorrhage as well as prevent thrombosis. The media and adventitia of the vessel wall enable vessels to dilate or constrict. The subendothelial basement membranes contain adhesive proteins which provide binding sites for platelet and leukocytes. Remodeling which occurs after injury is enhanced by extracellular matrix metalloproteinases.
Fibrinolysis
The fibrinolytic system provides a mechanism for removal of physiologically deposited fibrin. Clot lysis is brought about by the action of plasmin on fibrin. Fibrinolytic events are shown in Figure 15.2 . Plasminogen from circulating plasma is laid down with fibrin during the formation of thrombin. Plasminogen is primarily synthesized in the liver and circulates in two forms, one with a NH2-terminal glutamic acid residue (glu-plasminogen) and a second form with a NH2-terminal lysine, valine, or methionine residue (lys-plasminogen). Glu-plasminogen can be converted to lys-plasminogen by limited proteolytic degradation. Lys-plasminogen has a higher affinity for fibrin and cellular receptors; it is also more readily activated to plasmin than glu-plasminogen. Both forms of plasminogen bind to fibrin through specific lysine-binding sites. These lysine-binding sites also mediate the interaction of plasminogen with its inhibitor, α 2 -antiplasmin (α 2 AP). TAFI-mediated removal of C-terminal lysine and arginine residues will prevent high-affinity plasminogen binding and will attenuate fibrinolysis. Plasminogen is converted to its enzymatically active form, plasmin, by several activators. These activators are widely distributed in body tissues and fluids. t-PA is the principal intravascular activator of plasminogen. t-PA is a serine protease that binds to fibrin through lysine-binding sites. When t-PA is bound to fibrin its plasmin generation efficiency increases markedly. Urokinase (UK)-type plasminogen activator (u-PA), a second physiological activator of plasminogen, is present in urine and activates plasminogen to plasmin independent of the presence of fibrin. Plasmin splits fibrin and fibrinogen into fibrin-degradation products (FDPs): fragments X, Y, D, and E.
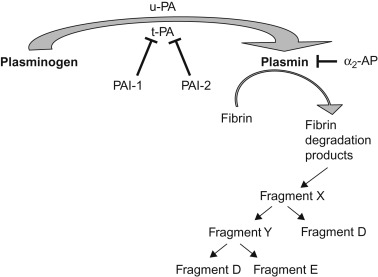
Properties attributed to the various fibrin split products include heparin-like effects, inhibition of platelet adhesion and aggregation, potentiation of the hypotensive effect of bradykinin and chemotactic properties (monocytes and neutrophils). Increased fibrinolysis is usually a reaction to intravascular coagulation (secondary fibrinolysis) rather than the initial event (primary fibrinolysis).
The action of plasmin is negatively regulated by several inhibitors (shown in Figure 15.2 ) these include α 2 AP and α 2 -macroglobulin. PA is in turn regulated by two inhibitors, plasminogen activator inhibitor-1 (PAI-1) and plasminogen activator inhibitor-2 (PAI-2). PAI-1 is the more physiologically important of these inhibitors.
Natural Inhibitors of Coagulation
In addition to the physiologic role of fibrinolysis, other inhibitors play critical roles in the control of hemostasis. Table 15.2 lists the plasma fibrinolytic components and hemostatic inhibitors and their principal substrates. All members of this group have overlapping roles in the control of coagulation and fibrinolysis. Major antiproteases of this group of inhibitors include antithrombin (AT), α 2 AP, α 2 -macroglobulin, the inhibitor of the activated first component of complement (C1 inhibitor), and α 1 -antitrypsin.
| Biologic half-life | Proteases inhibited | Concentration in plasma (mg/dl) | |
|---|---|---|---|
| FIBRINOLYTIC COMPONENTS | |||
| Plasminogen a | 48 h | — | 10–15 |
| PAs | |||
| Tissue | 3–4 min | — | — |
| Urokinase | 9–16 min | — | — |
| PAI a | — | PA, XII a | 60–200 b |
| INHIBITORS | |||
| AT a (heparin cofactor) | 17–76 h | XII a , XI a , IX a , X a , thrombin, kallikrein, plasmin | 10–14 104–121 b |
| α 2 -Plasmin inhibitor a (AP) | 30 h | XII a , XI a , kallikrein, plasmin, thrombin | 6–8 80–120 b |
| α 2 -Macroglobulin a | — | XII a , XI a , thrombin, kallikrein, plasmin | 190–310 |
| C1 inhibitor a | — | XII a , kallikrein | 20–25 |
| α 1 -Antitrypsin a | — | Thrombin, XI a , kallikrein | 245–325 |
| TAFI | 20–400 c | ||
| TFPI | — | Factor VII a /TF complex | Endothelial bound |
| Protein C a | 6 h | V a , VIII a , PAI | 0.4–0.6 |
| 71–109 b | |||
| Protein S | 60 h | V a , VIII a | 95–125 b |
| Protein C inhibitor a | — | Protein C a | 0.5 |
a Enzymatic activity: serine protease.
AT neutralizes the procoagulants thrombin, FIXa, Xa, and XIa ( Figure 15.3 ). When bound to circulating heparin or heparin sulfate on endothelial cells, AT undergoes a conformational change with a dramatic increase in this activity. TFPI is responsible for inactivation of the FXa/FVIIa/TF complex.
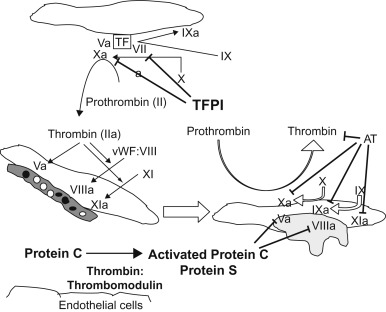
The vitamin-K-dependent zymogen, protein C, and its cofactor protein S, which is also a vitamin K protein, play an important role in the control of hemostasis by inhibiting activated factors V and VIII ( Figure 15.3 ). Binding of thrombin to thrombomodulin on endothelial cells of small blood vessels neutralizes the procoagulant activities of thrombin and activates protein C. Protein C (PC) binds to a specific receptor and the binding augments the activation of protein C by thrombin. Activated protein C inactivates factors Va and VIIIa in a reaction that is greatly accelerated by the presence of free protein S and phospholipids, thereby inhibiting the generation of thrombin. Free protein S itself has anticoagulant effects: it inhibits the prothrombinase complex (FXa, factor Va, and phospholipid), which converts prothrombin to thrombin and inhibits the complex of FIXa, factor VIIIa, and phospholipid, which converts factor X to FXa.
Hemostasis in the Newborn
Table 15.3 lists the hemostatic values in healthy preterm and term infants. In comparison with hemostatic mechanisms in older children and adults, those of newborn infants are not uniformly developed. The following physiological differences are present in the normal newborn infant:
| Normal adults/children | Preterm infant (28–32 weeks) | Preterm infant (33–36 weeks) | Term infant | |
|---|---|---|---|---|
| PT (s) | 10.8–13.9 | 14.6–16.9 | 10.6–16.2 | 10.1–15.9 |
| APTT (s) | 26.6–40.3 | 80–168 | 27.5–79.4 | 31.3–54.3 |
| Fibrinogen (mg/dl) | 95–425 | 160–346 | 150–310 | 150–280 |
| II (%) | 100 a | 16–46 | 20–47 | 30–60 |
| V (%) | 100 a | 45–118 | 50–120 | 56–138 |
| VII (%) | 100 a | 24–50 | 26–55 | 40–73 |
| VIII (%) | 100 a | 75–105 | 130–150 | 154–180 |
| vWF Ag (%) | 100 a | 82–224 | 147–224 | 67–178 |
| vWF (%) | 100 a | 83–223 | 78–210 | 50–200 |
| IX (%) | 100 a | 17–27 | 10–30 | 20–38 |
| X (%) | 100 a | 20–56 | 24–60 | 30–54 |
| XI (%) | 100 a | 12–28 | 20–36 | 20–64 |
| XII (%) | 100 a | 9–35 | 10–36 | 16–72 |
| XIII (%) | 100 a | — | 35–127 | 30–122 |
| PK (%) | 100 a | 14–38 | 20–46 | 16–56 |
| HMW-K (%) | 100 a | 20–36 | 40–62 | 50–78 |
a Expressed as a percentage of activity in pooled control plasma.
Plasma Factors:
In newborns, plasminogen levels are only 50% of adult values and α 2 AP levels are 80% of adult values, whereas PAI-1 and t-PA levels are significantly increased over adult values. The increased plasma levels of t-PA and PAI-1 in newborns on day 1 of life are in marked contrast to values from cord blood, in which concentrations of these two proteins are significantly lower than in adults. Newborns also have decreased activity of anticoagulant factors, especially AT, protein C, and protein S.
Blood vessels:
- •
Capillary fragility is increased.
- •
Prostacyclin production is increased.
Platelets:
- 1.
Platelet adhesion is increased due to increased vWF and increased HMW vWF multimers.
- 2.
Platelet aggregation abnormalities
- a.
Epinephrine-induced aggregation is decreased due to decreased platelet receptors for epinephrine.
- b.
Ristocetin-induced aggregation is increased due to increased vWF and increased HMW vWF multimers.
- a.
- 3.
Platelet activation is increased: as evidenced by elevated levels of thromboxane A2, B thromboglobulin, and PF4.
Detection of Hemostatic Defects
Evaluation of a patient for a hemostatic defect generally entails the following:
- 1.
Detailed history (see Table 15.4 for initial features suggestive of pathological bleeding in children)
- a.
Symptoms: Epistaxis, gingival bleeding, easy bruising, menorrhagia, hematuria, neonatal bleeding (heel stick, umbilicus), gastrointestinal (GI) bleeding, hemarthrosis, prolonged bleeding after lacerations.
- b.
Response to hemostatic challenge: Circumcision, surgery, phlebotomy, immunization/intramuscular injection, suture placement/removal, dental procedures.
- c.
Underlying medical conditions: Known associations with hemostatic defects (liver disease, renal failure, vitamin K deficiency).
- d.
Medications: Antiplatelet drugs (nonsteroidal anti-inflammatory drugs), anticoagulants (warfarin, heparin, low-molecular-weight heparin (LMWH)), antimetabolites ( l -asparaginase), prolonged use of antibiotics causing vitamin K deficiency, long-term use of iron suggestive of ongoing blood loss causing iron-deficiency anemia.
- e.
Family history: Symptoms, response to hemostatic challenge (siblings, parents, aunts, uncles, grandparents), transfusions after surgeries, iron deficiency after surgery or menorrhagia.
Table 15.4
Initial Features Suggestive of Pathologic Bleeding in Children
Age-related
Bleeding (e.g., umbilical stump bleeding, Intracranial hemorrhage, excessive and prolonged bleeding postcircumcision or after heel stick or intramuscular injection) during neonatal period
Palpable and multiple bruises in infants and older children who are not independently mobile
Persistent palpable bruising in an older mobile child
Spontaneous bleeding in the absence of anatomic causes
Personal history of
Recurrent (especially excessive and spontaneous) mucocutaneous bleeding
Atypical bleeds (e.g., hemarthroses, retroperitoneal bleeding), whether spontaneous or provoked
Excessive or prolonged bleeding after hemostatic challenges (i.e., trauma, dental procedures, or surgery)
Menorrhagia in adolescent girls: menstrual bleeding for >7 days or 80-ml blood loss per menstrual cycle (as evidenced by soaking through a pad or tampon within 1 h or change of pads or tampons every hour or passage of large (1.1-inch diameter) clots)
Traumatic bleeding that is out of proportion to or inconsistent with reported injury (consider nonaccidental trauma)
Family history of
Recurrent bleeding symptoms
Excessive or prolonged bleeding after trauma or invasive procedures
Known or suspected bleeding diatheses
Physical findings
Multiple bleeding stigmata
Physical findings suggestive of specific underlying causes (e.g., petechiae in platelet disorders, jaundice in liver disease, hypermobility, vascular malformations, musculoskeletal abnormalities)
Pallor/anemia
Reproduced with permission from Pediatrics , p. 883, Copyright © 2013 by the AAP.
- a.
- 2.
Complete physical examination
- a.
Signs consistent with past coagulopathy: Petechiae, ecchymosis, hematomas, synovitis/joint effusion, arthropathy, muscle atrophy, evidence of joint laxity or hyperextensibility which can exacerbate the bleeding phenotype. In very young patients parental joint mobility should be assessed.
- a.
- 3.
Laboratory evaluation ( Tables 15.1, 15.2, 15.3, and 15.5 ):
Table 15.5
Coagulation Tests and Normal Values
Test
Normal value
Clinical application
PLATELET FUNCTION
Template bleeding time (min)
<9
Crude, lack of reproducibility
Platelet aggregation
(see Chapter 14 )
Platelet Factor3 availability
Screens for platelet procoagulant activity
Clot retraction
Starts at hour 1; completes at hour 24
Measures platelet interaction with fibrin
INTRINSIC SYSTEM
Activated partial thromboplastin time(s)
25–35
EXTRINSIC SYSTEM
Prothrombin time(s)
10–12
Factor assays
See Tables 15.1 and 15.3
Thrombin time(s)
<24
Prolonged in hypofibrinogenemia, dysfibrinogenemia, hypoalbuminemia, liver disease, neonates
Reptilase time(s)
<25
Modification of thrombin time, unaffected by presence of heparin
ANTIPHOSPHOLIPID ASSAYS
Dilute Russell’s viper venom time(s)
29–42
Kaolin clotting time(s)
Sensitive test even in presence of heparin
FIBRINOLYTIC SYSTEM
Euglobulin clot lysis time (minutes)
90–240
Prolonged with hypofibrinolysis
Initial screening tests
- 1.
Complete blood count (CBC): Quantitative assessment of platelets and review of blood smear to assess platelet morphology.
- 2.
Assessments of platelet function:
- a.
Platelet Function Analyzer (PFA-100): Assesses flow through a membrane, membrane closure time is measured in response to ADP and to epinephrine. The closure time is often prolonged with impaired platelet function (see Chapter 14 ).
- b.
Bleeding time no longer used where PFA-100 is available because of difficulties in validation.
- a.
- 3.
Coagulation factor screening tests:
- a.
From a laboratory perspective the coagulation system is divided into the intrinsic pathway, the extrinsic pathway and the common pathway. Such an artificial division is not physiologically based but is useful for conceptualizing in vitro laboratory testing ( Figure 15.4 ).
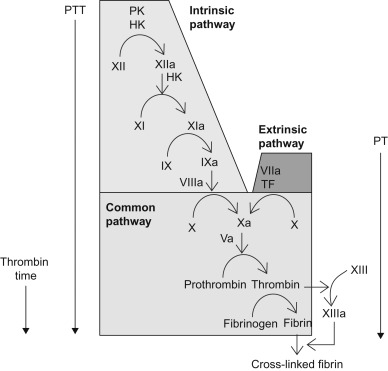
Figure 15.4
A conceptualization of commonly used screening tests of coagulation and the coagulation parameters they measure. PTT: partial thromboplastin time, PT: prothrombin time.
- a.
- 4.
Prothrombin time (PT) assay (assesses the extrinsic system): This test utilizes tissue thromboplastin and calcium chloride, to initiate the formation of thrombin via the extrinsic pathway. The international normalized ratio (INR) is used to correct for differences between thromboplastin potency across laboratories.
- 5.
Activated partial thromboplastin time (aPTT) assay (assesses the intrinsic system): This test utilizes a phospholipid reagent, a particulate activator (e.g., ellagic acid, kaolin, silica, soy extract) and calcium chloride to start the enzyme reaction that leads to the formation of thrombin via the intrinsic pathway.
Common Confirmatory Coagulation Assays
- –
Fibrinogen: Quantitative measurement of fibrinogen, useful when both the PT and the aPTT are prolonged.
- –
Thrombin time: Prolonged when fibrinogen is reduced or abnormal, in the presence of inhibitors (FDPs, d -dimers) and in the presence of thrombin-inhibiting drugs and hypoalbuminemia. This is a useful test to diagnose dysfibrinogenemia (qualitatively abnormal fibrinogen). Also useful when both the PT and aPTT are prolonged. The reptilase time is a modification of the TT using the purified enzyme reptilase instead of thrombin and is unaffected by heparin and heparin-like anticoagulants.
- –
Mixing studies (performed to evaluate a prolonged PT or aPTT): The respective assay is performed following addition of normal pooled plasma to patient plasma. Normalization indicates a clotting factor deficiency that was corrected by addition of normal pooled plasma. Continued prolongation indicates presence of a coagulation inhibitor. Such inhibitors may be physiologically relevant or only detected in vitro .
- –
Clotting factor activity assays: Performed to identify clotting factor deficiencies if mixing studies normalize. FXII, FXI, FIX, and FVIII assays are useful if the aPTT normalizes in mixing studies. The FVII assay is useful if the PT normalizes in mixing studies. FX, FV, FII, and fibrinogen assays are useful if both PT and aPTT normalize in mixing studies. ( Note : Factor XIII deficiency does not result in prolongation of the PT or aPTT.)
- –
von Willebrand antigen: Quantitative assay for vWF, useful when PFA-100 closure time is prolonged or when von Willebrand disease (vWD) is suspected.
- –
vWF (ristocetin cofactor activity): Functional/qualitative assay for vWF, useful when the PFA-100 closure time is prolonged or when vWD is suspected.
- –
vWF multimers are useful when discrepancies between vWF antigen (normal to low) and ristocetin cofactor activity (extremely low; ratio of ristocetin cofactor activity to vWF antigen <0.6).
- –
Platelet aggregation studies: A qualitative assessment of platelet function, useful when platelet function disorders are suspected or PFA-100 closure time is prolonged.
- –
Urea clot lysis assay: Useful screening for FXIII deficiency outside the newborn period although not sensitive enough to make an accurate diagnosis. Fetal fibrinogen interferes with this assay giving a false-positive result in the newborn period. In the absence of fibrin crosslinkage by FXIII, a clot will degrade with incubation in 5M urea. As this assay detects only severe factor XIII deficiency, functional FXIII assays should be conducted if mild deficiency is suspected. FXIII activity, antigen, and FXIII A and B subunit sequencing are essential to make an accurate diagnosis as per recommendations of the International Society of Thrombosis and Hemostasis.
Global Hemostatic Tests
Hemostasis is a complex interplay of simultaneously occurring events, with current assays only reflecting snapshots of this dynamic process. Global hemostatic tests can provide detailed information on thrombin generation and processes downstream including fibrin polymerization and fibrin dissolution.
- –
Thrombin generation assay: The Calibrated Automated Thrombogram (CAT) system uses a fluorogenic substrate to continuously measure the generated thrombin. The endogenous thrombin potential (ETP), which can be measured by calculating the area under the curve from the thrombogram, has shown correlation with the bleeding phenotypes in hemophilia, in hemophilia patients with inhibitors, and factor XI deficiency.
- –
Thromboelastography (TEG): TEG is performed on whole blood, assessing the viscoelastic property of clot formation under low shear condition after the addition of specific coagulation activators. Improvement in the methodology is widely expanding the use of TEG from preclinical research settings to point of care testing in intensive care units and operating rooms. The device has a metal pin suspended by a torsion wire immersed into a cup which holds the whole blood. Once clotting starts, fibrin strands formed increase the torque between the pin and the cup which is measured electronically. TEG provides various data relating to clot formation and fibrinolysis (the lag time before the clot starts to form, the rate at which clotting occurs, the maximal amplitude of the trace or clot strength, and the extent and rate of amplitude).
Preoperative Evaluation of Hemostasis
- 1.
History
The history is perhaps the most important element of the evaluation. In an effort to standardize bleeding histories, a number of quantitative bleeding assessment tools (BATs) have been developed and validated in individuals with known vWD (Vicenza-based BAT, the condensed MCMDM-1 VWD, ISTH, and the more pediatric-specific PBQ), although whether these tools can directly predict future bleeding episodes needs further study.
- a.
If negative: No coagulation tests are indicated; only a CBC.
- b.
If positive or unreliable: The following tests should be performed: CBC, PFAs, PT, aPTT, and fibrinogen, TT, von Willebrand panel.
- a.
- 2.
Abnormal tests require further investigation (see Figure 15.5 ).
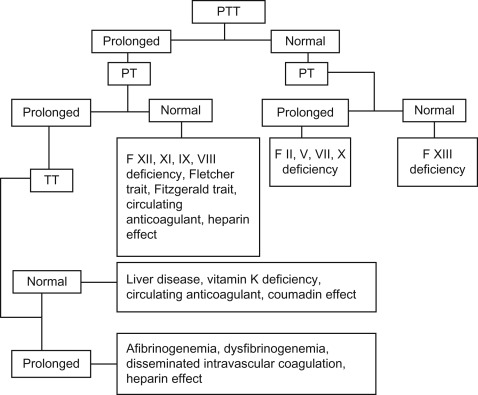
Figure 15.5
Coagulation tests and interpretation. PTT: activated partial thromboplastin time, PT: prothrombin time, TT: thrombin time.
- 3.
In a patient with a significant bleeding history, if all screening tests and von Willebrand panel are normal consider FXIII, PAI-1 activity, α 2 AP, and platelet aggregation studies, vitamin C levels and rule out hyperextensibility on clinical examination.
Acquired Coagulation Factor Disorders
Vitamin K Deficiency
The normal full-term infant is born with levels of factors II, VII, IX, and X that are low by adult standards ( Table 15.3 ). The coagulation factors fall even lower over the first few days of life, reaching their nadir on about the third day. This is due to the low body stores of vitamin K at birth. As little as 25 µg vitamin K can prevent this fall in activity of the vitamin-K-dependent clotting factors. The vitamin K content of cow’s milk is only about 6 µg/dl and that of breast milk 1.5 µg/dl. Moreover, breastfed infants are colonized by lactobacilli that do not synthesize gut vitamin K. It is a combination of low initial stores and subsequent poor intake of vitamin K that occasionally produces an aggravation of the coagulation defect causing primary hemorrhagic disease of the newborn. Vitamin K deficiency results in hemorrhagic disease between the second and fourth days of life and is manifested by GI hemorrhage, hemorrhage from the umbilicus, or internal hemorrhage (classic hemorrhagic disease of the newborn). Bleeding attributable to this cause is responsive to parenteral vitamin K therapy; for this reason parenteral vitamin K is routinely administered to newborns. Serious occurrences of vitamin K deficiency bleeding continue to occur due to parental refusal of vitamin K prophylaxis after birth and increased prevalence of home deliveries with the aid of midwives who may not be licensed to administer intramuscular vitamin K after birth.
In premature infants of low birth weight, both the vitamin K stores and the level of coagulation factors are even lower than in term infants. The response to vitamin K is slow and inconsistent, suggesting that the immature liver has reduced synthetic capability.
Maternal ingestion of certain drugs may result in neonatal hypoprothrombinemia and reduction in factors VII, IX, and X, resulting in early hemorrhagic disease of the newborn. These drugs include oral anticoagulants and anticonvulsants (phenytoin, primidone, and phenobarbital). This can be prevented by administration of vitamin K to the mother 2–4 weeks prior to delivery.
Late hemorrhagic disease of the newborn occurs between day 7 and 6 months of life in the absence of adequate vitamin K prophylaxis at birth. It may be idiopathic or exacerbated by malabsorption or liver disease. Consider investigating for biliary atresia, alpha 1 antitrypsin deficiency, malabsorption conditions like celiac disease, cystic fibrosis if hemorrhagic disease of the newborn occurs after adequate vitamin K prophylaxis at birth.
Table 15.6 lists the laboratory findings in vitamin K deficiency in relationship to the findings in liver disease and disseminated intravascular coagulation (DIC).
| Component | Vitamin K deficiency | Liver disease | DIC |
|---|---|---|---|
| Red cell morphology | Normal | Target cells | Fragmented cells, burr cells, helmet cells, schistocytes |
| aPTT | Prolonged | Prolonged | Prolonged |
| PT | Prolonged | Prolonged | Prolonged |
| Fibrin split products | Normal | Normal or slightly increased | Markedly increased |
| Platelets | Normal | Normal | Reduced |
| Factors decreased | II,VII, IX, X | I, II, V, VII, IX, X | Assays are of limited utility |
Hepatic Dysfunction
Any transient inability of the newborn liver to synthesize necessary coagulation factors, even in the presence of vitamin K, can result in hemorrhagic disease that is nonresponsive to vitamin K therapy. Hepatic dysfunction as a result of immaturity, infection, hypoxia, or underperfusion of the liver can all result in transient inability of the liver to synthesize coagulation factors. This is more prominent in small premature infants. The sites of bleeding in these cases are usually pulmonary and intracerebral, with a high mortality. Other causes of hepatocellular dysfunction affecting patients of all ages, include hepatitis, cirrhosis, Wilson disease, and Reye syndrome. In liver disease vitamin-K-dependent factors, FV, and fibrinogen are usually decreased, fibrin split products may be elevated due to impaired clearance ( Table 15.6 ). In contrast, factor VIII levels are usually normal. There is no response to vitamin K. There is usually a clinical response to clotting factor replacement therapy, using fresh frozen plasma (FFP) and cryoprecipitate (replacement guidelines are the same as those outlined in Table 15.7 ).
|
a One bag of cryoprecipitate contains about 200 mg fibrinogen.
Disseminated Intravascular Coagulation
DIC is characterized by the intravascular consumption of platelets and plasma clotting factors. Widespread coagulation within the vasculature results in the deposition of fibrin thrombi and the production of a hemorrhagic state when the rapid utilization of platelets and clotting factors results in levels inadequate to maintain hemostasis. The accumulation of fibrin in the microcirculation leads to mechanical injury to the red cells, resulting in erythrocyte fragmentation and microangiopathic hemolytic anemia.
Widespread activation of the coagulation cascade rapidly results in the depletion of many clotting factors as fibrinogen is converted to fibrin throughout the body as follows:
- 1.
The generation of thrombin results in intravascular coagulation, rapidly falling platelet count, fibrinogen, and FV, FVIII, and FXIII levels. Paradoxically, in vitro bioassays for these factors may be elevated owing to generalized activation of the coagulation system.
- 2.
Concurrently, plasminogen is converted to its enzymatic form (plasmin) by t-PA. Plasmin digests fibrinogen and fibrin (secondary fibrinolysis) into fibrin split products (FSPs), resulting in clot lysis.
Diagnosis of DIC relies on the presence of a well-defined clinical situation associated with a thrombo-hemorrhagic disorder. Table 15.6 lists the typical diagnostic findings in DIC. Disease states associated with DIC and low-grade DIC are listed in Table 15.8 . Generally available treatment options for treatment of DIC are shown in Table 15.7 .
| Causative factors | Clinical situation |
|---|---|
| Tissue injury | Trauma/crush injuries |
| Head injury | |
| Major surgery | |
| Heat stroke | |
| Burns | |
| Venoms | |
| Malignancy | |
| Obstetrical accidents | |
| Amniotic fluid embolism | |
| Placental abruption | |
| Stillborn fetus | |
| Abortion | |
| Fat embolism | |
| Endothelial cell injury | Infection (bacterial, viral, protozoal) |
| AND/OR | Immune complexes |
| Abnormal vascular surfaces | Eclampsia |
| Postpartum renal failure | |
| Oral contraceptives | |
| Cardiopulmonary bypass | |
| Giant hemangioma | |
| Vascular aneurysm | |
| Cirrhosis | |
| Malignancy | |
| Respiratory distress syndrome | |
| Platelet, leukocyte, or red cell injury | Incompatible blood transfusion |
| Infection | |
| Allograft rejection | |
| Hemolytic syndromes | |
| Drug hypersensitivity | |
| Malignancy | |
| Localized intravascular coagulopathy | Kasabach–Merritt syndrome |
| Chronic inflammatory disorders | |
| Arteriovenous fistulae | |
| Vascular prosthesis | |
| Glomerulonephritis |
Low-grade DIC has the potential to accelerate into fulminant DIC. Careful monitoring in terms of detecting the presence of fragmented red blood cells, mild decrease in platelets and low fibrinogen levels usually indicate high fibrinolysis and increased risk of bleeding. Treatment of low-grade DIC involves treating the underlying disease triggering it.
Inherited Coagulation Factor Disorders
Table 15.9 lists the genetics, prevalence, coagulation studies, and symptoms of inherited coagulation factor disorders.
| Factor deficiency | Genetics | Estimated prevalence | Prevalence of ICH (upper limits) | APTT | PT | Associated with bleeding episodes |
|---|---|---|---|---|---|---|
| Afibrinogenemia | AR | 1:500,000 | 10% | P | P | ++ |
| Dysfibrinogenemia | AR | 1:1 million | Single case | N/P | P | +/− thrombosis |
| II | AR | 1:1 million | 11% | P | P | ++ |
| V (parahemophilia) | AR | 1:1 million | 8% of homozygotes | P | P | ++ |
| VII | AR | 1:300,000 | 4–6.5% | N | P | + |
| VIII (hemophilia A) | XLR | 1:5000 males | 5–12% | P | N | +++ |
| von Willebrand’s disease | 1:1000 | Extremely rare | ||||
| Type 1 | AD | N/P | N | + | ||
| Type 2 | AD | N/P | N | ++ | ||
| Type 3 | AR | P | N | ++ | ||
| IX (hemophilia B) | XLR | 1;25,000 males | 5–12% | P | N | +++ |
| X | AR | 1:1 million | 21% | P | N | ++ |
| XI (hemophilia C) | AV | 1:100,000 | Extremely rare | P | N | + |
| XII | AD | P | N | − | ||
| XIII | AR | 1:2 million | 33% | N | N | + a |
| Prekallikrein (Fletcher trait) | AD | P b | N | − | ||
| HMW kininogen (Fitzgerald trait) | AR | P | N | − | ||
| Passovoy (?) | AR | P | N | +/− |
a Umbilical stump bleeding; need FXIII activity and gene sequencing for FXIII A and B subunits for accurate diagnosis. The 5M urea lysis solubility test is not a sensitive test for diagnosis.
Hemophilia A and B
The most common coagulation disorders after vWD are hemophilia A and B. Hemophilia A is an X-linked recessive bleeding disorder attributable to decreased blood levels of functional procoagulant factor VIII (FVIII, VIII: C, antihemophilic factor). Hemophilia B is also an X-linked recessive disorder and is indistinguishable from hemophilia A with respect to its clinical manifestations. In hemophilia B, the defect is a decreased level of functional procoagulant factor IX (FIX, IX: C, plasma thromboplastin component or Christmas factor). The incidence of hemophilia is probably 1 per 5,000 live male births with hemophilia B being one-fifth as common as hemophilia A. Thus factor VIII deficiency accounts for 80–85% of cases of hemophilia, with factor IX deficiency accounting for the remainder. Given X-linked recessive inheritance, both types occur with similar incidence among all races and in all parts of the world.
Hemophilia A Carrier Detection
Excessive lyonization may result in reduced FVIII levels in female carriers of hemophilia; hence a reduced FVIII level can have utility in diagnosing the carrier state. However, a normal FVIII level does not rule out carrier status, which is most accurately diagnosed by genetic testing.
Direct gene mutation analysis: The FVIII common intron 22 inversion, resulting from an intrachromosomal recombination, is identifiable in 45% of severe hemophilia A patients. For the remaining 55% of patients with severe hemophilia A, as well as all those with mild and moderate hemophilia A, the molecular defects can usually be detected by efficient screening of all 26 FVIII exons and splice junctions. Therefore, targeted mutation analysis is the most accurate test for carrier detection and prenatal diagnosis for severe hemophilia A. For rare patients in whom a precise mutation cannot be identified or gene sequencing is not an option, intragenetic and extragenetic linkage analysis of DNA polymorphisms can be useful with up to 99.9% precision (when an affected male patient and his related family members are available). Preimplantation genetic diagnosis is a reproductive option available to carrier females.
When definitive diagnosis of the carrier state cannot be made, determination of the FVIII/vWF:Ag ratio (<1.0) can be used to detect 80% of hemophilia A carriers with 95% accuracy. Use of this methodology requires careful standardization of the laboratory performing the testing.
Hemophilia B Carrier Detection
Hemophilia B carriers have a wide range of FIX levels but, in a subset of cases, can be detected by the measurement of reduced plasma factor IX activity (in 60–70% of cases).
Targeted mutation analysis: The factor IX gene is located centromeric to the factor VIII gene in the terminus of the long arm of the X chromosome. There is no linkage between the FVIII and FIX genes. The 34 kb FIX coding sequence comprises eight exons and encodes a 461-amino-acid precursor protein that is approximately one-third the size of the factor VIII cDNA. Because of the smaller gene size, FIX mutations can be identified in nearly all patients. Direct FIX mutation testing is available through DNA diagnostic laboratories, with linkage analysis used in those cases where the responsible mutation cannot be identified.
Prenatal Diagnosis
Prenatal diagnosis of hemophilia can be performed by either chorionic villus sampling (CVS) at 10–12 weeks gestation or by amniocentesis after 15 weeks gestation. If DNA analysis is not available or if a woman’s carrier status cannot be determined, fetal blood sampling can be performed at 18–20 weeks gestation for direct fetal factor VIII plasma activity level measurement. The normal fetus at 18–20 weeks gestation has a very low FIX level, which an expert laboratory can distinguish from the virtual absence of FIX in a fetus with severe hemophilia B.
Maternal–fetal combined complication rates for amniocentesis and CVS are 0.5–1.0% and 1.0–2.0%, respectively. Fetal blood sampling is less available; the fetal loss rate for these procedures ranges from 1% to 6%.
Clinical Course of Hemophilia
Hemophilia should be suspected when unusual bleeding is encountered in a male patient. Clinical presentations of hemophilia A and hemophilia B are indistinguishable. The frequency and severity of bleeding in hemophilia are usually related to the plasma levels of factor VIII or IX ( Table 15.10 ), although some genetic modifiers of hemophilia severity have been identified. The median age for first joint bleed is 10 months, corresponding to the age at which the infant becomes mobile. Table 15.11 shows the common sites of hemorrhage in hemophilia. The incidence of severity and clinical manifestations of hemophilia are listed in Table 15.12 .
| Type | Percentage factor VIII/IX | Type of hemorrhage |
|---|---|---|
| Severe | < 1 | Spontaneous; hemarthroses and deep soft tissue hemorrhages |
| Moderate | 1–5 | Gross bleeding following mild to moderate trauma; some hemarthrosis; seldom spontaneous hemorrhage |
| Mild | 5–40 | Severe hemorrhage only following moderate to severe trauma or surgery |
| High-risk carrier females | Variable | Gynecologic and obstetric hemorrhage common, other symptoms depend on plasma factor level. |
|
| Severity | Severe | Moderate | Mild |
|---|---|---|---|
| INCIDENCE | |||
| Hemophilia A | 70% | 15% | 15% |
| Hemophilia B | 50% | 30% | 20% |
| BLEEDING MANIFESTATIONS | |||
| Age of onset | ≤1 year | 1–2 years | 2 years–adult |
| Neonatal hemorrhages | |||
| Following circumcision | Common | Common | None |
| Intracranial | Occasionally | Rare | Rare |
| Post Neonatal period | |||
| Muscle/joint hemorrhage | Spontaneous | Following minor trauma | Following major trauma |
| CNS hemorrhage | High risk | Moderate risk | Rare a |
| Postsurgical hemorrhage | Common | Common | Rare a |
| Oral hemorrhage b | Common | Common | Rare a |
For hemophilia B, the Leyden phenotype (severe hemophilia as a child that becomes mild after puberty) has been described in families with defects in the androgen-sensitive promoter region of the gene.
Treatment (Factor Replacement Therapy)
Factor replacement therapy is the mainstay of hemophilia treatment. The degree of factor correction required to achieve hemostasis is largely determined by the site and nature of the particular bleeding episode. Commercially available products for replacement therapy are listed in Table 15.13 . Commercially available FVIII products include high-purity recombinant preparations, highly purified plasma-derived concentrates (monoclonal/immunoaffinity purified) and intermediate-purity plasma-derived preparations. Available FIX products include recombinant FIX and plasma-derived high-purity FIX concentrate (coagulation FIX concentrate). Recently recombinant products with the FVIII or FIX product fused to the Fc portion of immunoglobulin 1 (Ig1) have become available which prolong the half-life of the factor in circulation. This enables prophylaxis dosing up to 3–5 days for FVIII-deficient patients and up to weekly dosing for FIX-deficient patients. New products in the pipeline include PEGylation of the factor molecule, which by increasing the molecular mass, reduces glomerular filtration, proteolytic degradation, and clearance. The combined effect is to increase the half-life of the PEGylated protein. Several other strategies being investigated include single-chain rFVIII which more effectively binds to vWF, bioengineered antibodies, and short interfering RNAs which slow down the endogenous AT synthesis. Gene therapy offers the promise of curing hemophilia by inserting the deficient gene into the patient’s tissue which can then permanently restore circulating factor levels. Moreover, the gene for FIX is small and easy to insert into many vectors.
| Class | Product | Purity | Procedure | Primary use |
|---|---|---|---|---|
| FVIII Plasma Derived | Koate-DVI (Bayer Corp) | Intermediate purity | SD/Dry Heat | Hemophilia A |
| Humate P (Aventis Behring) | Intermediate purity | P | Hemophilia A/vWD | |
| Alphanate SD-HT (Grifols Corp) | High purity | SD/Dry Heat | Hemophilia A /vWD | |
| Hemofil M (Baxter Bioscience) | Ultra high purity | SD | Hemophilia A | |
| Monoclate P (Aventis Behring) | Ultra high purity | P | Hemophilia A | |
| FVIII Recombinant | 1 st generation – Recombinate (Baxter Bioscience) | Ultra high purity | None | Hemophilia A |
| 2 nd generation – Helixate (Aventis Behring) | Ultra high purity | SD | Hemophilia A | |
| 2 nd generation – ReFacto (Wyeth) | Ultra high purity | SD | Hemophilia A | |
| 2 nd generation – Kogenate FS (Bayer Corp) | Ultra high purity | SD | Hemophilia A | |
| 3 rd generation – Advate (Baxter Bioscience) | Ultra high purity | SD | Hemophilia A | |
| 3 rd generation – Xyntha (Wyeth) | Ultra high purity | SD | Hemophilia A | |
| FVIII Fc Fusion Long Acting | Eloctate (Biogen) | High | SD/Nanofiltration | Hemophilia A |
| FIX Plasma Derived | Alphanine SD-VF (Grifols Corp) | High | SD/nanofiltration | Hemophilia B |
| Mononine (Aventis Behring) | High | SD/nanofiltration | Hemophilia B | |
| FIX Recombinant | BeneFIX (Wyeth) | High | Nanofiltration | Hemophilia B |
| FIX Fc Fusion Long Acting | Alprolix (Biogen) | High | Nanofiltration | Hemophilia B |
| Activated PCC | FEIBA VH (Basxter Bioscience) | N/A | Vapor heat | Inhibitor bypass therapy |
| PCC | Profilnine SD (Grifols Corp) | Intermediate | SD | Rare bleeding disorders |
| Bebulin VH (Baxter Bioscience) | Intermediate | Vapor heat | Rare bleeding disorders | |
| Proplex T (Baxter Bioscience) | Intermediate | Dry heat | FVII deficiency in places where recombinant product not available. | |
| Bypassing agent: | NovoSeven (NovoNordisk) | N/A | None | Inhibitor bypass therapy, FVII deficiency |
| Specialty items | Kcentra (CSL Behring) | Ion exchange | Anticoagulant reversal |
In 2011 Nathwani and colleagues successfully transduced six hemophilia B patients with all of them experiencing stable FIX expression at 1–7% of normal levels. The same group has shown long-term therapeutic factor IX expression in 10 patients associated with clinical improvement and no late toxic effects when assessed for a period of up to 3 years.
Prothrombin complex concentrates (PCCs), formerly a mainstay of hemophilia B treatment, are not utilized because of the risk of thrombotic complications associated with intensive treatment. Source plasma for all plasma-derived factor concentrates undergoes donor screening and nucleic acid testing for a variety of viral pathogens. In addition all plasma-derived and many recombinant factor concentrates undergo a viral inactivation treatment, typically with solvent detergent, wet or dry heat treatment, pasteurization or nanofiltration. Factor concentrates are preferred over FFP or cryoprecipitate because the plasma-derived concentrates undergo viral inactivation treatment in addition to donor screening. Recombinant factor concentrates are widely accepted as the treatment of choice for previously untreated patients, minimally treated patients, and patients who have not had transfusion-associated infections.
Strategies for hemophilia care include on-demand treatment of acute bleeding episodes or, for severe hemophilia patients, prophylactic administration of clotting factor concentrate to maintain trough factor levels >1% augmented with on-demand treatment of breakthrough bleeding episodes. The latter strategy pharmacologically converts the severe hemophilia phenotype to a moderate phenotype with an attendant reduction in frequency of bleeding episodes. A randomized multicenter US national study suggested a markedly reduced incidence of hemophilic arthropathy when prophylaxis was instituted prior to onset of recurrent joint bleeds. This benefit must be balanced with the need for frequent prophylactic infusions (3–4 times/week for FVIII, 2 times/week for FIX), venous access considerations, potential requirements for central venous access devices, increased cost of treatment, and the occasional patients who have a very mild clinical course. Table 15.14 provides generally accepted guidelines for treatment of most types of hemophilic bleeding. When a bleeding episode is suspected hemostatic treatment should be rendered first then diagnostic evaluation(s) may be performed.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


