Introduction
Human growth is an astonishing process. Its beginnings are intertwined with the enormously intricate mechanisms that transform a single cell into a complex embryo. Once formed, the human fetus and then child continue to grow over the course of approximately 15 years, reaching a body mass more than 10,000,000 times greater than that of the zygote from which the child originated. The early growth rate is enormous. The fetus grows in length at more than 100 cm per year, but, by birth, the linear growth rate has slowed to 50 cm per year and by midchildhood to 5 cm per year. This decline in the growth rate is briefly interrupted by the pubertal growth spurt but then resumes, causing body growth to gradually grind to a halt as the child approaches adulthood. During the period of body enlargement, growth of the different organs, such as the kidney, heart, lung, liver, and bones, occurs synchronously, orchestrated to maintain body proportions.
In the majority of children, the largely mysterious mechanisms governing body growth proceed like clockwork, yielding an adult height most often between 1.5 and 2 m. However, in some children, body growth is either greater than or less than the normal range, often prompting the concerned family to seek medical evaluation.
The healthcare provider is then called upon to answer two questions. First, why is the child’s growth outside the normal range and second what, if anything, should be done about it. The first question, “why,” is important for several reasons. Many children are short or tall because they have inherited from their parents a large set of normal polymorphisms that place the child in one tail of the normal height distribution or the other. Such polygenic short or tall stature is generally benign. However, some children are short or tall because of a single gene abnormality that may have important health implications, such as a propensity to cardiac malformations or to malignancies. Other children are short because of a subtle systemic acquired disease, such as inflammatory bowel disease (IBD), autoimmune thyroiditis, or a central nervous system (CNS) tumor causing pituitary dysfunction. Thus the healthcare provider who evaluates children’s growth faces the challenge of sorting out the few children with important underlying pathology from the many children with a benign condition.
The second question, “what to do about it,” can also be challenging. If the diagnostic evaluation uncovers an underlying, treatable disorder, such as celiac disease, the course is clear. But often, no simple treatable cause is found. Then, the provider and the family must try to assess whether the short or tall stature is sufficiently abnormal and sufficiently distressing to the child, or likely to be distressing in the future, so that the possible benefits of medical therapy outweigh the effort, costs, discomforts, and potential risks of intervening.
Addressing these two central questions requires a deep understanding of normal childhood growth and the many disorders that affect it, the determination to look diligently for underlying causes, as well as the willingness and ability to listen compassionately to the concerns of the child and family. In this chapter, we have tried to present the healthcare provider with a broad overview of the knowledge necessary for this endeavor.
Childhood growth
The Biological Basis of Linear Growth
In children, many tissues and organs grow simultaneously. However, most children who present to the pediatric endocrinologist for evaluation of growth do so because the child’s height is abnormal. The height is primarily determined by the length of the long bones in the lower extremities and the height of the vertebral bodies. These bone lengths in turn are determined by the action of the skeletal growth plates, thin layers of cartilage located near the ends of the long bones and vertebrae.
Within the growth plates, the chondrocytes are arranged in columns parallel to the long axis of the bone ( Fig. 11.1A ). In the upper portion (closer to the ends of the bone) of the columns, the chondrocytes proliferate ( Fig. 11.1B ). In the lower portion of the columns, the chondrocytes stop dividing and instead physically enlarge ( Fig. 11.1B ). These two processes, cell proliferation and cell hypertrophy, lead to chondrogenesis, the production of cartilage. In isolation, this chondrogenesis would cause progressive thickening of the cartilaginous growth plate. However, simultaneously, at the bottom of the growth plate, the newly formed cartilage is remodeled into bone. The net result is that new bone tissue is progressively created at the bottom of the growth plates, the overall bone grows longer, and the child grows taller.
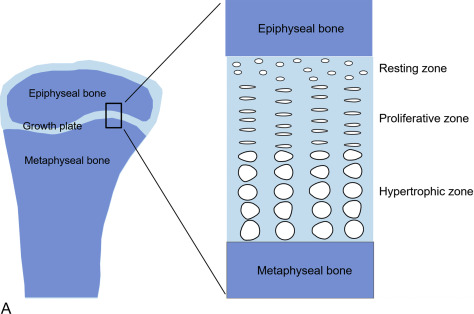
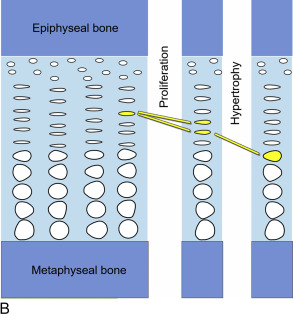
Thus linear growth (height gain) in children results from growth plate chondrogenesis. Therefore short stature is caused by decreased growth plate chondrogenesis and tall stature is caused by increased growth plate chondrogenesis.
Regulation of Linear Growth
Because linear growth results from growth plate chondrogenesis, the regulation of linear growth results from the regulation of growth plate chondrocytes. Factors that stimulate growth plate chondrocyte proliferation and hypertrophy stimulate linear growth, whereas factors that inhibit growth plate chondrocyte proliferation and hypertrophy inhibit linear growth. This regulation of growth plate chondrocyte function occurs at multiple different levels; endocrine signals, inflammatory cytokines, nutritional intake, paracrine/autocrine signals, extracellular matrix effects, and intracellular systems can all modulate chondrocyte proliferation and hypertrophy and therefore affect the rate of linear growth. Consequently, abnormalities at any of these levels can cause short or tall stature.
Endocrine Regulation of Linear Growth
Growth Hormone
The Pituitary Gland
The pituitary gland lies in the sella turcica , the hypophyseal fossa of the sphenoid bone, which is located in the center of the cranial base. The concept of the pituitary as a “master gland” controlling the endocrine activities of the body has become outdated and has been replaced by an appreciation of the importance of the brain, particularly of the hypothalamus, in regulating hormonal production and secretion. Nevertheless, the pituitary gland remains central to our understanding of the regulation of growth, metabolism and homeostasis, response to stress, lactation, and reproduction.
Embryologically, the pituitary gland is formed from two distinct sources, namely Rathke’s pouch, a diverticulum of the primitive oral cavity (stomodeal ectoderm), which gives rise to the adenohypophysis, and the neural ectoderm of the floor of the forebrain, which gives rise to the neurohypophysis, the posterior lobe. The adenohypophysis normally constitutes 80% of the weight of the pituitary and consists of the pars distalis (also known as the pars anterior or anterior lobe ), the pars intermedia (also known as the intermediate lobe ), and the pars tuberalis (also known as the pars infundibularis or pars proximalis ).
Much of our knowledge of normal hypothalamopituitary development is derived from animal, particularly rodent, models. In the mouse, a thickening of the ectoderm in the midline of the anterior neural ridge, forming the hypophyseal placode, heralds the onset of pituitary development at 7.5 dpc (days postcoitum). The formation of a rudimentary Rathke’s pouch follows at 9 dpc, with formation of a definitive pouch by 12 dpc and subsequently, the anterior pituitary consisting of five different cell types secreting six different hormones ( Fig. 11.2 ).
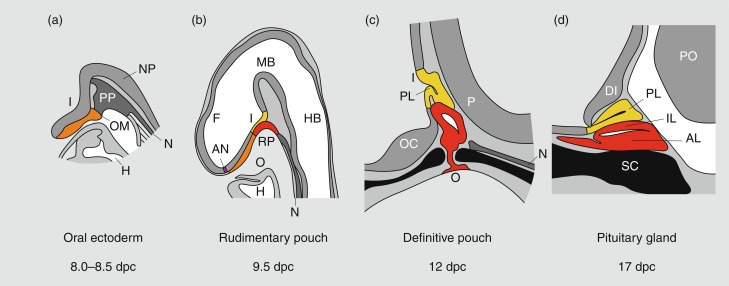
In humans, the pars distalis is the largest portion of the adenohypophysis and houses the great majority of hormone-producing cells. In contrast with the mouse, the pars intermedia is rudimentary and consists of several cystic cavities lined by a single layer of cuboidal epithelium as it largely disappears during embryogenesis. The pars distalis and intermedia are separated by a cleft, a vestigial structure of Rathke’s pouch from which it develops. This structure may often develop as a cyst (Rathke’s cleft cyst). The pars tuberalis represents an upward extension of the pars distalis onto the pituitary stalk and may contain a limited number of gonadotropin-producing cells. The posterior pituitary (neurohypophysis) consists of the infundibular stem or hypophyseal stalk, the median eminence of the tuber cinereum, and the infundibular process (posterior lobe, neural lobe). The posterior pituitary contains the terminal axonal projections of magnocellular neurons from the paraventricular and supraoptic nuclei of the hypothalamus; these produce oxytocin, which is required during lactation and parturition, and vasopressin, which is required for osmotic regulation. It has no known function in the regulation of growth and will not be discussed further in this chapter.
Rathke’s pouch, the origin of the adenohypophysis, can be identified in the 3-mm embryo during the third week of pregnancy in humans. Rathke’s pouch then begins to develop, resulting in a complete pouch disconnected from the oral ectoderm by the end of the sixth gestational week. Growth hormone (GH)-producing cells can be identified by 9 weeks of gestation. It is at about this time that the vascular connections between the anterior lobe of the pituitary and the hypothalamus develop, although it has been demonstrated that hormonal production by the pituitary can occur in the absence of connections with the hypothalamus. Somatotropes are thus frequently demonstrable in the pituitary of an anencephalic newborn. Nevertheless, it appears likely that the initiation of development of the anterior pituitary is dependent on responsiveness of the oral ectoderm to inducing factors from the ventral diencephalon. Infrequently, the craniopharyngeal canal (marking the embryonic migration of Rathke’s pouch) remains patent and may contain small nests of adenohypophyseal cells—giving rise to a pharyngeal hypophysis that may be capable of hormone synthesis.
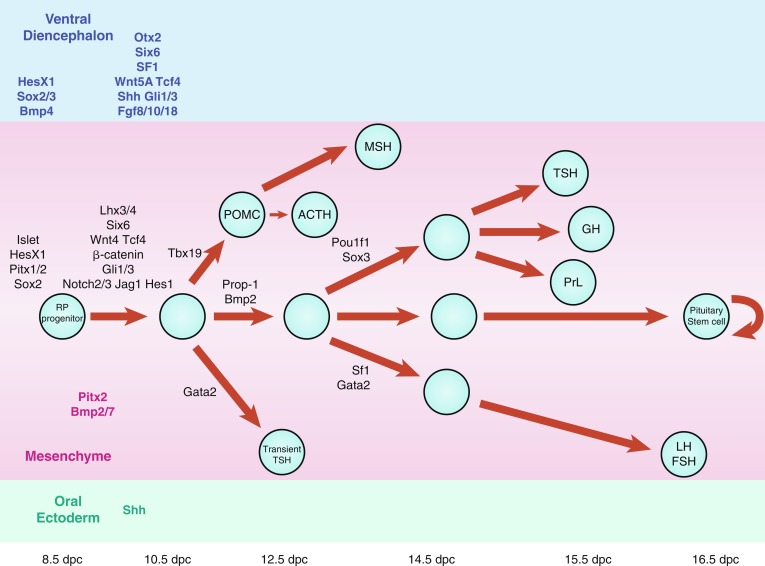
Maintained apposition and interaction between the oral ectoderm and neuroectoderm is critical for normal anterior pituitary development. Experimental manipulation of embryos from several species, as well as Rathke’s pouch explant experiments in rodents, have shown that signals from the diencephalon are essential not only for the induction and maintenance of Rathke’s pouch, but also for the regionalization within the pouch that allows the emergence of the different endocrine cell types. During gestation, proliferating progenitor cells are enriched around the pouch lumen, and they appear to delaminate as they exit the cell cycle and differentiate. During late mouse gestation and the postnatal period, anterior lobe progenitors reenter the cell cycle and expand the populations of specialized, hormone-producing cells. At birth, all cell types are present, and their localization appears stratified, based on cell type. Current models of cell specification in the anterior lobe suggest that opposing gradients of fibroblast growth factor (FGF) and bone morphogenic protein (BMP) signaling pattern the progenitor cells within Rathke’s pouch before they move on to the anterior lobe where they differentiate. Several studies have revealed that normal pituitary development is dependent upon a complex cascade of transcription factors and signaling molecules that are expressed in a spatiotemporal manner.
Signaling molecules implicated in pituitary development are either intrinsic, emanating from the oral ectoderm, such as sonic hedgehog (Shh), or extrinsic from the neuroectoderm, such as Nkx2.1, FGFs (e.g., FGF 8), and BMPs (e.g., BMP4) ( Fig. 11.3 ). These molecules may activate or repress transcription factors, such as Hesx1, Lhx3, and Lhx4. They may also act as morphogens creating the appropriate environment for cell differentiation, thus playing a critical role in cell fate. Such signaling molecules include members of the Shh family, FGFs, transforming growth factors-β (TGFs)/BMPs, Wingless/Wnts, and molecules in the Notch pathway to mention a few.
Recently, Davis and colleagues have challenged the current dogma of pituitary cell specification, showing evidence that, in mice, the pattern of cell specification that results in the rostral location of gonadotropes, the caudal location for somatotropes and a more intermediate location for corticotropes and thyrotropes, does not appear to be the result of an ordered cell cycle exit, as previously suggested. All anterior lobe cell types appear to begin the differentiation process concurrently (E11.5-E14.5), rather than in a temporally discrete manner.
To date, not many pituitary phenotypes have been reported in association with mutations in these signaling molecules. Importantly, the Wnt signaling pathway has recently been implicated in pituitary tumorigenesis. For example, there is clear evidence that the Wnt/β catenin pathway is involved in the pathogenesis of craniopharyngioma, a rare tumor in the hypothalamopituitary region.
Multiple pituitary-specific transcription factors are involved in the determination of pituitary cell lineages and cell-specific expression of anterior pituitary hormones (see Fig. 11.3 ). Several homeodomain transcription factors have been shown to be involved in human anterior pituitary development and differentiation. Defects in several of these have now been associated with various combinations of pituitary hormone deficiencies ( Table 11.1 ). Because additional gene defects have been implicated in abnormal murine hypothalamopituitary development, it seems likely that the number of known human genetic defects will expand.
| Gene | Pituitary Deficiencies | MRI Phenotype | Inheritance | Other Phenotypic Features |
|---|---|---|---|---|
| POU1F1 | GH, TSH, prolactin | Small or normally sized anterior pituitary | AR and AD | |
| PROP1 | GH, TSH, LH, FSH, prolactin, evolving ACTH deficiencies | Small, normal or enlarged anterior pituitary – may evolve over time | AR | |
| HESX1 | Isolated GH deficiency through to panhypopituitarism with TSH, LH, FSH, ACTH, and prolactin deficiencies | Optic nerve hypoplasia, absence of the septum pellucidium, ectopic posterior pituitary, anterior pituitary hypoplasia | AR and AD | Developmental delay, visual abnormalities, SOD |
| LHX3 | GH, TSH, LH, FSH, prolactin with late ACTH deficiencies | Small, normal, or enlarged anterior pituitary | AR | Short neck with limited rotation |
| LHX4 | GH, TSH, ACTH, and gonadotrophin deficiencies | Small anterior pituitary, ectopic posterior pituitary, cerebellar abnormalities, corpus callosum hypoplasia | AD (variable penetrance) | |
| SOX3 | GH, TSH, LH, FSH, ACTH deficiencies. Most commonly isolated GH deficiency | Anterior pituitary and infundibular hypoplasia, ectopic posterior pituitary, corpus callosum abnormalities including cysts; persistent craniopharyngeal canal | X-linked recessive | Learning difficulties |
| SOX2 | LH, FSH variable GH deficiency | Anterior pituitary hypoplasia, optic nerve hypoplasia, SOD, hypothalamic hamartoma | AD (usually de novo) | Microphthalmia, anophthalmia, micropenis, sensorineural deafness, gastrointestinal tract defects with esophageal atresia |
| GLI2 | GH, TSH, and ACTH with variable gonadotrophin deficiencies | Anterior pituitary hypoplasia | AD | Holoprosencephaly, cleft lip and palate, anophthalmia, postaxial polydactyly, imperforate anus, laryngeal cleft, renal agenesis |
| GLI3 | GH, TSH, LH, FSH, ACTH | Anterior pituitary hypoplasia | AD | Pallister-Hall syndrome Postaxial polydactyly, hamartoma |
| OTX2 | GH, TSH, LH, FSH, and ACTH deficiencies | Normal or small AP, pituitary stalk agenesis, ectopic posterior pituitary, Chiari I malformation | AD (usually de novo) | Microcephaly, bilateral anophthalmia, developmental delay, cleft palate |
| FGFR1 | GH, TSH, LH, FSH, and ACTH deficiencies | Normal or small anterior pituitary, corpus callosum agenesis | AD | ASD and VSD, brachydactyly, brachycephaly, preauricular skin tags, ocular abnormalities, seizures |
| FGF8 | GH, TSH, ACTH, gonadotrophin, and ADH deficiencies | Absent corpus callosum, optic nerve hypoplasia, holoprosencephaly | AD or AR | |
| PROKR2 | GH, TSH, ACTH and gonadotrophin deficiencies | Hypoplastic corpus callosum, normal or small anterior pituitary | AD | |
| ARNT2 | GH, TSH, ACTH, LH, FSH, ADH deficiencies | Absent PP, thin stalk, thin corpus callosum, delayed myelination | AR | Hip dysplasia, hydronephrosis, vesicoureteric reflux, neuropathic bladder, microcephaly, prominent forehead, deep-set eyes, retrognathia |
| TCFL7 | GH | Absent posterior pituitary, anterior pituitary hypoplasia, optic nerve hypoplasia, partial agenesis of corpus callosum, thin anterior commissure | AD | |
| IGSF1 RNPC3 KCNQ1 | GH (transient/partial), TSH, prolactin GH deficiency GH, ACTH, TSH, and gonadotrophin deficiencies | Normal in the majority of cases. Frontoparietal hygroma, hypoplasia of the corpus callosum, and small stalk lesion reported Small anterior pituitary with normal stalk and eutopic posterior pituitary Normal/small AP | X-linked recessive AR AR | Macroorchidism, delay in puberty Maternally inherited gingival fibromatosis |
In the human adult, the pituitary has a mean weight of 600 mg, with a range of 400 to 900 mg. Pituitary weight is slightly greater in women than in men, and typically increases during puberty and pregnancy. In the newborn, pituitary weight averages about 100 mg. The pituitary normally resides in the sella turcica immediately above and partially surrounded by the sphenoid bone. The volume of the sella turcica provides a good measure of pituitary size, which may be reduced in the child with pituitary hypoplasia. It is important, however, to recognize that considerable variation in pituitary size occurs normally. The pituitary is covered superiorly by the diaphragma sellae, and the optic chiasm is directly above the diaphragma. The anatomic proximity between the optic chiasm and the pituitary is important because hypoplasia of the optic chiasm may occur together with hypothalamic/pituitary dysfunction, as in the condition of septooptic dysplasia (SOD), and because pituitary tumors may in turn impact on the optic chiasm, leading to visual impairment. The patient with congenital blindness or nystagmus should be initially evaluated and then subsequently monitored carefully for hypopituitarism, as this can evolve. In addition, suprasellar growth of a pituitary tumor may initially manifest with visual complaints or evidence of progressive impairment of peripheral vision, particularly bitemporal hemianopia.
The existence of a portal circulatory system within the pituitary is critical to normal pituitary function. The blood supply of the pituitary derives from the superior and inferior hypophyseal arteries, branches of the internal carotid. The anterior and posterior branches of the superior hypophyseal artery may terminate within the infundibulum and the proximal portion of the pituitary stalk. Hypothalamic peptides, produced in neurons that terminate in the infundibulum, enter the primary plexus of the hypophyseal portal circulation and are transported by means of the hypophyseal portal veins to the capillaries of the anterior pituitary. This portal system thus provides a means of communication between the neurons of the hypothalamus and the hormone-producing cells of the anterior pituitary. The blood supply of the neurohypophysis is separate, deriving from the inferior hypophyseal artery. Regulation of the posterior lobe of the pituitary does not involve the hypophyseal portal circulation but, rather, is mediated through direct neural connections.
The definitive Rathke’s pouch comprises proliferative progenitors that will gradually relocate ventrally, away from the lumen as they differentiate. A proliferative zone containing progenitors is maintained in the embryo in the perilumenal area and was recently found to persist in the adult. The exact nature of progenitor cells in the pituitary gland, however, remains unknown. Members of the Sox family of transcription factors are likely involved in the earliest steps of pituitary stem cell proliferation and the earliest transitions to differentiation. The transcription factor Prophet of Pit1 (PROP1) and the NOTCH signaling pathway may then regulate the transition to differentiation. It has been proposed that the niche of progenitors may be the marginal zone around the lumen of Rathke’s pouch, between the anterior and intermediate lobes of mouse pituitary, because cells in this region are able to give birth to all five pituitary hormone cell lineages (see Fig. 11.3 ).
Stem cells have been shown to play a role in tumorigenesis in some tissues, and their role in pituitary hyperplasia, pituitary adenomas, and tumors is an important area for future investigation. The ability to cultivate and grow pituitary stem cells in a predifferentiation state might also be helpful for the long-term treatment of pituitary deficiencies. Indeed, a seminal study resulted in the efficient self-formation of a three-dimensional anterior pituitary in an aggregate culture of mouse embryonic stem (ES) cells. Various endocrine cells were generated, and these cells were able to respond to trophic hormones. When these ES-derived cell aggregates were implanted under the kidney capsule in hypophysectomized mice, corticosterone was produced. These studies may therefore reflect the first step toward stem cell treatment.
Terminally differentiated secreting cells are not distributed randomly in a patchwork-like fashion throughout the pituitary gland. Instead, these cells appear to organize themselves in same-cell–type networks. The connectivity between the cells of this network is important to deliver coordinated secretory pulses of hormones to their target tissues and to facilitate the coordinated physiological response to stimuli.
Growth Hormone Chemistry
Human GH is produced from somatotrope cells within the anterior pituitary as a single-chain, nonglycosylated, 191-amino-acid, 22-kDa protein that comprises a core of four helices in a parallel/antiparallel orientation with two disulfide bonds between cysteines 53–165 and 182–189. The 217-amino-acid GH precursor is cleaved to remove the signal peptide before secretion.
GH is homologous with several other proteins produced by the pituitary or placenta, including prolactin, chorionic somatomammotropin (CS, placental lactogen), and a 22-kDa GH variant hGH-V, secreted only by the placenta. The latter differs from pituitary GH by 13 amino acids. The genes for these proteins have probably descended from a common ancestral gene, even though the genes are now located on different chromosomes.
GH1 is located on the long arm of chromosome 17 (17q22-24) within a cluster of five homologous genes encompassing a distance of about 65 kb ( CSHP [CS pseudogene], CSH-1 [CS gene], GH-2 and CSH-2 ). Expression of GH1 is regulated by the highly polymorphic proximal promoter and a locus control region (LCR) 15–32 kb upstream of the gene that confers the pituitary-specific and high-level expression of GH. Normally, the vast majority of GH (75%) produced by the pituitary is of the mature 22-kDa form. Alternative splicing results in deletion of amino acids 32 through 46, yielding a 20-kDa form that normally accounts for less than 10% of pituitary GH. The remainder of pituitary GH includes desamidated and N-acetylated forms, as well as various GH oligomers. A 17.5-kDa variant that results from complete skipping of exon 3 and lacks amino acids 32–71 is much less abundant (1%–5%).
Growth Hormone Secretion
GH can be identified in fetal serum by the end of the first trimester. The pulsatile pattern characteristic of GH secretion largely reflects the interplay of multiple regulators, including two hypothalamic regulatory peptides: GH-releasing hormone (GHRH) and somatostatin (somatotropin release–inhibiting factor [SRIF]). GHRHR encodes a G-protein–coupled receptor comprising seven transmembrane domains. The expression of GHRHR is upregulated by pituitary-specific transcription factor 1 ( POU1F1 ) and is required for the proliferation of somatotrophs.
Regulation of GH production by GHRH is largely transcriptionally mediated and is dependent on stimulation of adenylate cyclase and increases in intracellular cyclic adenosine monophosphate (cAMP) concentrations. Solid tumors secreting GHRH are a rare cause of GH excess. GHRH is used diagnostically, especially for the identification of adult GH deficiency, when it is frequently used in combination with arginine. However, it is not conventionally used for therapeutic purposes in patients with GH deficiency.
The actions of the 14-amino-acid peptide somatostatin appear to regulate the timing and amplitude of pulsatile GH secretion, rather than GH synthesis. The binding of somatostatin to its specific receptor results in an inhibition of adenylate cyclase activity and a reduction in intracellular calcium concentrations. The pulsatile secretion of GH observed in vivo is believed to result from a simultaneous reduction in hypothalamic somatostatin release and increase in GHRH activity. Conversely, a trough of GH secretion occurs when somatostatin release is increased in the presence of diminished GHRH activity. The net effects of these two hypothalamic hormones is modulation of GH secretion, as well as the timing and amplitude of peaks, resulting in pulsatile GH secretion. Multiple neurotransmitters and neuropeptides are involved in regulating the release of these hypothalamic factors, including serotonin, histamine, norepinephrine, dopamine, acetylcholine, gamma-aminobutyric acid (GABA), thyroid-releasing hormone, vasoactive intestinal peptide, gastrin, neurotensin, substance P, calcitonin, neuropeptide Y, vasopressin, corticotrophin-releasing hormone, and galanin. These factors are implicated in the alterations of GH secretion observed in a wide variety of physiological states (such as stress, sleep, hemorrhage, fasting, hypoglycemia, and exercise) and form the basis for a number of GH-stimulatory tests used in the evaluation of GH secretory capacity/reserve. GH secretion is also impacted by a variety of nonpeptide hormones, including androgens, estrogens, thyroxine, and glucocorticoids. The precise mechanisms by which these hormones regulate GH secretion are complex, potentially involving actions at the hypothalamic and pituitary levels. Practically speaking, hypothyroidism and glucocorticoid excess may each blunt spontaneous and provocative GH secretion (and therefore should be corrected before GH testing). Sex steroids, at the onset of puberty or administered pharmacologically, appear to be responsible for the rise in GH secretion characteristic of puberty. Synthetic hexapeptides capable of stimulating GH secretion have been developed and termed GH-releasing peptides (GHRPs). These peptides, later recognized as analogues of the gastric hormone ghrelin, are capable of directly stimulating GH release and enhancing the GH response to GHRH. These agents have the potential advantage of oral administration, and, in the patient with an intact pituitary, may be capable of greatly enhancing GH secretion. When these agents were administered chronically to elderly patients and to some GH-deficient children, the amplitudes of GH pulses were significantly increased. Ghrelin-mimetic ligands were used to characterize a common receptor termed the GH secretagogue receptor (GHS-R) for the GH-releasing substances. The GHS-R is a G-protein–coupled receptor, which is distinct from the GHRH receptor. The receptor is strongly expressed in the hypothalamus, but specific binding sites for GHRPs have also been identified in other regions of the CNS and peripheral endocrine and nonendocrine tissues in both humans and other organisms. Ghrelin is a 28-amino-acid peptide that has been identified as the endogenous ligand for GHS-R. It is expressed predominantly in the stomach, but smaller amounts are also produced within the bowel, pancreas, kidney, the immune system, placenta, pituitary, testis, ovary, and hypothalamus. Ghrelin is a unique gene product that requires octanoylation for normal function. Intravenous, intracerebroventricular, and intraperitoneal administration of ghrelin in animal models stimulates food intake and obesity and raises plasma GH concentrations, and, to a lesser extent, prolactin (PRL) and adrenocorticotropic hormone (ACTH) concentrations. In addition, it influences endocrine pancreatic function and glucose metabolism, gonadal function, and behavior. It also controls gastric motility and acid secretion and has cardiovascular and antiproliferative effects. Both ghrelin and GHRPs release GH synergistically with GHRH but the efficacy of these compounds as growth-promoting agents is poor. Variants in the ghrelin receptor have been identified as a possible cause of idiopathic short stature (ISS) and GH deficiency. However, note that mouse models with targeted deletion of the receptor ( ghsr -/- ) have a near normal phenotype. A second peptide encoded by the same gene as ghrelin has been identified and termed obestatin . This gene appears to regulate weight but not GH secretion.
In addition to the complex regulatory processes described previously, the synthesis and secretion of GH are also regulated by feedback by the insulin-like growth factor (IGF) polypeptides. IGF receptors have been identified in the pituitary. Inhibition of GH secretion by IGF-1 has been demonstrated in multiple systems. In addition, inhibition of spontaneous GH secretion has been demonstrated in humans treated with subcutaneous injections of recombinant IGF-1. GH also regulates its own secretion by acting directly on hypothalamic GH receptors (GHRs), via a short loop negative feedback.
During puberty, sex steroids increase GH pulse amplitude contributing to the very high serum concentrations of IGF-1 characteristic of puberty. GH secretion begins to decline by late adolescence and continues to fall throughout adult life. Indeed, puberty may be considered with some justification a period of “acromegaly,” whereas aging (with its characteristic decrease in GH secretion) has been termed the somatopause.
In addition to aging, a wide variety of physiological conditions affect GH secretion. These include stage of sleep, nutritional status, acute fasting, exercise, stress, and sex steroids. Ho and associates have reported that neither age nor sex influenced the integrated serum concentrations of GH when the effects of estradiol were removed from analysis. The effects of testosterone on serum IGF-1 concentrations may be at least in part independent of GH because individuals with mutations of the GHR still experience a rise in serum IGF-1 during puberty.
The pulsatile nature of GH secretion is readily demonstrable by frequent serum sampling, especially when coupled with sensitive assays for GH. Such assays demonstrate that, under normal conditions, serum GH concentrations are less than 0.2 ng/mL between bursts of GH secretion. It is consequently impractical to assess GH secretion by a single, random serum sample. Maximal GH secretion occurs during the night, especially at the onset of the first slow-wave sleep (stages III and IV). Rapid-eye-movement (REM) sleep is, on the other hand, associated with low GH secretion.
Normal young men generally experience, on average, 12 GH secretory bursts per 24 hours. Obesity is characterized by decreased GH secretion, reflected by a decreased number of GH secretory bursts. Fasting increases the number and amplitude of GH secretory bursts, presumably reflecting decreased somatostatin secretion. The impact of the pulsatile secretory nature of GH secretion on its biological actions remains uncertain.
Growth Hormone Receptor/Growth Hormone-Binding Protein
The GHR comprises an extracellular, hormone-binding domain, followed by a single membrane-spanning domain, and a cytoplasmic domain. Two genomic GHR isoforms that exist only in humans have arisen from ancestral homologous recombination. They differ in the retention or deletion of exon 3. Exon 3 of the GHR has been shown to be deleted in a number of normal individuals. This delta-3 GHR polymorphism has been shown by some, but not all, investigators to affect responsiveness to GH and to be associated with birth size and postnatal growth. There are also short isoforms of the GHR because of alternative splicing, resulting in a truncated receptor with loss of a large portion of the cytoplasmic domain.
The GHR is a member of the class 1 hematopoietic cytokine family and is highly homologous with the prolactin receptor and shares sequence homology with many of the receptors for interleukins (ILs), as well as receptors for erythropoietin (EPO), leptin, granulocyte-macrophage colony-stimulating factor, and interferon.
Examination of the crystal structure of the GH-GHR complex revealed that the complex consists of one molecule of GH bound to two GHR molecules, indicating a GH-induced receptor dimerization—which is necessary for GH action. Interestingly, a genetically engineered fusion complex of GH and the GHR has been shown to have a significantly improved efficacy and a dramatically longer half-life compared with GH alone when tested in rodent models.
The GHR, like its family group member EPO-R, is preformed as a dimer and is transported in a nonligand bound state to the cell surface. GH then binds in a sequential manner to the GHR dimer where the first GHR binds to the stronger site 1 of the GH molecule followed by the second GHR binding to the weaker site 2. Binding of GH results in a conformational change whereby rotation of the GHRs results in repositioning of the intracellular domains and of Box11-1–associated Janus Kinase 2 (JAK2), a major GHR-associated tyrosine kinase. As a result, JAK2 is autophosphorylated and activated, which in turn leads to cross-phosphorylation of distal tyrosine residues of GHR. This enables SH2 ( Src homology 2) domain molecules to dock to these sites. The GHR itself appears to have no intrinsic kinase activity. It is likely that colocalization of two JAK2 molecules by the dimerized GHR results in transphosphorylation of one JAK2 by the other, leading to JAK2 activation. Signal transducer and activator of transcription (Stat)5a and 5b molecules contain SH2 domains and bind to these phosphorylated tyrosine sites, and then they in turn become phosphorylated. Phosphorylated Stat5 molecules (homo- and hetero-) dimerize and translocate to the nucleus where they bind deoxyribonucleic acid (DNA), as dimers or as tetramers, and activate target genes. Importantly, the dimerization of the GHR by GH is exploited by the use of Pegvisomant, a genetically modified form of GH, which binds to one receptor only and therefore cannot dimerize the receptor. It therefore acts as a GH antagonist and can be used to treat pituitary gigantism and acromegaly.
Both Stat5a and Stat5b can be activated by GH, and they have both overlapping and distinct functions. Gene inactivation mouse models have shown that Stat5b is of greater importance for stimulation of growth than Stat5a.
Negative Regulation of Growth Hormone Signaling
Activation of JAK-STAT signaling occurs rapidly, within minutes after GH stimulation, but is transient because of the tight control of the termination of signaling. This negative regulation of signaling occurs at several levels: GHR internalization, suppressors of cytokine signaling (SOCS), protein tyrosine phosphatases (PTPs), and protein inhibitors of activated Stats (PIAS).
In humans, the circulating GH-binding protein (GHBP) appears to derive from proteolytic cleavage of the extracellular domain of the GHR. GHBP has an identical affinity for GH as GHR. It binds to GH with high specificity and affinity, but with relatively low capacity. It increases GH half-life in the circulation and may serve a function in the transportation of GH to target tissues and subsequent binding to the receptor. GHBP is, like GHR, present in many tissues, but GHBP in the circulation is mostly derived from the liver. Although GHR and GHBP are regulated by and are very sensitive to GH, and GHR and GHBP often change in parallel, measurement of plasma GHBP has not been shown to reflect GHR and GH responsiveness. Measurement of serum GHBP concentrations may, however, aid in identifying patients with GH insensitivity (GHI) caused by genetic abnormalities of the GHR. Patients with GHI from nonreceptor abnormalities, abnormalities of the intracellular portion of the GHR, or inability of the receptor to dimerize may have normal serum concentrations of GHBP. Recently, a complex of the GH and the GHBP molecule has been shown to be more effective than GH alone—indicating a physiological and possible therapeutic role for GHBP.
Growth Hormone Actions
According to the somatomedin hypothesis, the anabolic actions of GH are mediated through the IGF polypeptides. Although this hypothesis is at least in part true, it appears that GH is capable of stimulating a variety of effects that are independent of IGF activity. Indeed, the effects of GH and IGF are on occasion contradictory, as for example, the “diabetogenic” actions of GH and the glucose-lowering activity of IGFs. Green and colleagues attempted to resolve some of these differences in a “dual-effector” model in which GH stimulates precursor cells, such as prechondrocytes, to differentiate. When differentiated cells or neighboring cells then secrete IGFs, these peptides act as mitogens and stimulate clonal expansion. This hypothesis is based on the ability of IGF peptides to work not only as classic endocrine factors that are transported through the blood but as paracrine or autocrine growth factors. GH also stimulates a variety of metabolic effects, some of which appear to occur independently of IGF production, such as lipolysis, amino acid transport in diaphragm and heart, and production of specific hepatic proteins. Thus there are multiple sites of GH action, and it is not entirely clear which of these actions are mediated through the IGF system and which might represent IGF-1–independent effects of GH.
Insulin-Like Growth Factor-1
Insulin-Like Growth Factor-1 Historical Background
The IGFs (or somatomedins) constitute a family of peptides that are at least in part GH-dependent and that are believed to mediate many of the anabolic and mitogenic actions of GH. Although they were originally identified in 1957 by their ability to stimulate [35S] sulfate incorporation into rat cartilage, it has been established over the ensuing 45 years that they are involved in diverse metabolic activities.
Insulin-Like Growth Factor Structure and Molecular Biology
IGF-1, previously termed somatomedin-C , is a basic polypeptide of 70 amino acids, whereas IGF-2 is a slightly acidic polypeptide of 67 amino acids. The two peptides are structurally related, sharing 45 of 73 possible amino acid positions. They have approximately 50% amino acid homology to insulin. Like insulin, both IGFs have A and B chains connected by disulfide bonds and an intervening connecting (C-peptide) region. This structural homology explains the ability of both IGFs to bind to the insulin receptor and for insulin to bind to the type 1 IGF receptor. On the other hand, structural differences explain the failure of insulin to bind to the IGF-binding proteins.
Serum Concentrations of Insulin-Like Growth Factors
In human fetal serum, IGF-1 concentrations are relatively low and are positively correlated with gestational age. A correlation between fetal cord serum IGF-1 concentrations with birth weight has been reported by some groups, although others have reported no correlation. IGF-1 concentrations in human newborn serum are generally 30% to 50% of adult concentrations. There is a slow, gradual rise in serum concentrations during childhood, with attainment of adult concentrations at the onset of sexual maturation. During puberty, IGF-1 concentrations increase to 2 to 3 times the concentrations seen in adults. Thus concentrations during adolescence correlate better with Tanner stage (bone age) than with chronological age. Girls with gonadal dysgenesis show no adolescent increase in serum IGF-1, clearly establishing the association of the pubertal rise in IGF-1 with the production of sex steroids. Estrogen stimulates GH secretion, which increases hepatic IGF-1 production. It is of note, however, that patients with GHI because of GHR mutations show a pubertal rise in serum IGF-1, which may be mediated by direct effects of androgen on IGF-1.
After adolescence, or at least after 20 to 30 years of age, serum IGF-1 concentrations demonstrate a gradual and progressive age-associated decline. It has been suggested that this decline may be responsible for the negative nitrogen balance, decrease in body musculature, and osteoporosis characteristic of aging. Although this provocative hypothesis remains unproven at this time, it has generated considerable interest in the potential use of GH and/or IGF-1 therapy in normal aging.
Human newborn concentrations of IGF-2 are generally 50% of adult concentrations. By 1 year of age, however, adult concentrations are attained with little, if any, subsequent decline even out to the seventh or eighth decade of life. This is in contrast to rodents, whose IGF-2 expression declines early following birth. Lack of rodent models has impeded our understanding of the physiological role of IGF-2 persistence in humans.
Insulin-Like Growth Factor Receptors
IGFs bind (although generally with low affinity) to insulin receptors, thus providing an explanation for their insulin-like activity. In addition, IGFs bind to at least two classes of IGF receptors.
The type 1 IGF receptor is closely related to the insulin receptor. Both are heterotetramers composed of two identical membrane-spanning alpha subunits and two identical intracellular beta subunits. The alpha subunits contain the binding sites for IGF-1 and are linked by disulfide bonds. The beta subunits contain a transmembrane domain, an adenosine triphosphate (ATP)-binding site, and a tyrosine kinase domain, which constitutes the presumed signal transduction mechanism for the receptor.
Although the type 1 IGF receptor has been commonly referred to as the IGF-1 receptor , the receptor is capable of binding both IGF-1 and IGF-2 with high affinity—and both IGF polypeptides appear capable of activating tyrosine kinase by binding to this receptor. Affinity of the type 1 receptor for insulin is generally 100-fold less, thereby providing one of the mechanisms for the mitogenic effects of insulin. IGF-1 acts primarily through IGF1R but can bind with lower affinity to the highly homologous insulin receptor (IR), and to IGF1R/IR heterodimers. Vice versa, insulin is able to signal through the IGF1R. Signs of such alternative signaling can become apparent in pathological IGF-1 or insulin signaling.
The type 1 IGF receptor mediates IGF actions on multiple cell types, and these actions are diverse and tissue specific. In general, it is believed that all of the effects of IGF receptor activation are mediated by tyrosine kinase activation and phosphorylation of substrates, which activate specific cellular pathways, leading to various biological actions. Among these effects is induction of cell growth through activation of the cell cycle machinery, maintenance of cell survival (prevention of apoptosis) mediated by effects on the Bcl family members, and induction of cellular differentiation, which occurs by as yet incompletely characterized mechanisms.
The substrates phosphorylated by the IGF receptor include members of the insulin receptor substrate family (particularly IRS-1 and IRS-2); knockout of these genes results in poor growth in mice (as well as insulin resistance). In addition, several other signaling molecules respond to IGF receptor activation. Blockade of the type I receptor has been proposed as a cancer therapy.
Mutations that decrease IGF signaling lead to an extension of life expectancy in nematodes, flies, and mice. It is unclear, however, what relevance the IGF-1/IR has for human longevity. Recent data have suggested that loss-of-function variants in IGF1R are overrepresented among female centenarians, suggesting a role of this pathway in modulation of human life span.
The type 2 IGF receptor, however, bears no structural homology with either the insulin or the type 1 IGF receptor. The type 2 receptor does not contain an intrinsic tyrosine kinase domain or any other recognizable signal transduction mechanism and has been found to be identical to the cation-independent mannose-6-phosphate (CIM6P) receptor, a protein involved in the intracellular lysosomal targeting of a variety of acid hydrolases and other mannosylated proteins. Unlike the type 1 IGF receptor, which binds both IGF polypeptides with high affinity and insulin with 100-fold lower affinity, the type 2 receptor only binds IGF-2 with high affinity. IGF-1 binds with substantially lower affinity, and insulin not at all. Most studies have indicated that the classic mitogenic and metabolic actions of IGF-1 and IGF-2 are mediated through the type 1 IGF receptor, with its tyrosine kinase signal transduction mechanism.
Insulin-Like Growth Factor-Binding Protein Superfamily
Although insulin and the IGFs share significant structural homology, and despite the structural-functional similarity of the insulin and type 1 IGF receptors, the IGFs differ from insulin in that the IGFs circulate in plasma complexed to a family of binding proteins. These carrier proteins extend the serum half-life of the IGF peptides, transport the IGFs to target cells, and modulate the interaction of the IGFs with their surface membrane receptors. Six distinct IGFBPs have been identified. Additional lower affinity IGF binding proteins (named IGFBP related proteins , IGFBPrPs ) were found by in silico searches for homology to the known IGFBPs; many of these molecules were previously known in other contexts, serving roles in normal or neoplastic growth.
Under most conditions, the IGFBPs appear to inhibit IGF action—presumably by competing with IGF receptors for IGF polypeptides. This concept is supported by the observation that IGF analogues with decreased affinity for IGFBPs generally appear to have increased biological potency. In addition, IGFBP-3 appears to inhibit cell growth even in the absence of added IGF—suggesting a direct inhibitory role of the binding protein. Under specific conditions, however, several of the IGFBPs apparently are capable of enhancing IGF action—perhaps by facilitating IGF delivery to target receptors.
Evidence indicates that IGFBPs are essentially bioactive molecules that, in addition to binding IGFs, have a variety of IGF-1–independent functions. These include growth inhibition in some cell types, growth stimulation in other tissues, direct induction of apoptosis, and modulation of the effects of other non-IGF growth factors. These effects of IGFBPs are mediated by binding to their own receptors. Because IGFBP-3 is regulated by GH, it is intriguing that in vivo, IGFBP-3 enhances IGF-1 action when given to hypophysectomized rats (rather than inhibiting it). The mechanisms involved in this effect have not been elucidated but may explain the limited effects of IGF-1 therapy on the growth of Laron patients.
The relative amounts of each of the IGFBPs vary among biological fluids. IGFBP-1 is the major IGFBP in human amniotic fluid. IGFBP-2 is prominent in cerebrospinal fluid and seminal plasma. IGFBP-3 is the major IGFBP in normal human serum and demonstrates clear GH dependence. Among the IGFBPs, IGFBP-3 and IGFBP-5 are unique in that they normally circulate in adult serum as part of a ternary complex consisting of IGFBP-3 or IGFBP-5, an IGF polypeptide, and an acid-labile subunit (ALS).
Analysis of IGFBPs is further complicated by the presence of IGFBP proteases, capable of various levels of IGFBP degradation. Initially reported in the serum of pregnant women, proteases for IGFBP-3, -4, and -5 have already been demonstrated in a variety of biological fluids. Proteolysis of IGFBPs complicates their assay by both Western ligand blotting and radioimmunoassay methodologies and must be taken into consideration when concentrations of the various IGFBPs in biological fluids are reported. The physiological significance of limited proteolysis of IGFBPs remains to be determined, although evidence suggests that protease activity results in decreased affinity of the IGFBP for IGF peptides. Recently, recessive mutations in the metalloproteinase pregnancy-associated plasma protein A2 ( PAPP-A2 ) were associated with short stature. The mutations lead to increased binding of IGF-1 within the ternary complex and reduced free IGF-1.
Targeted Disruption of Components of the Insulin-Like Growth Factor System
The critical role of the IGF system in fetal and postnatal growth was demonstrated in a series of elegant gene knockout studies in mice. Unlike GH and GHR knockouts, which are near normal size at birth, Igf1 null mice have a birth weight 60% of normal. Postnatal growth is also abnormal. A similar prenatal and postnatal growth phenotype has been observed in a reported human case of an Igf1 gene deletion. Igf2 null mice also show diminished pre- and postnatal growth. The Igf2 gene is imprinted in humans, such that only the paternal gene is expressed. Therefore loss-of-function mutations on the paternal allele cause growth impairment whereas mutations on the maternal allele do not. When the gene for the type 1 IGF receptor is knocked out (Igf1r null mice), the mice are severely growth retarded. In humans, dominant and recessive IGF1R mutations are associated with intrauterine growth retardation (IUGR), postnatal growth failure, and learning difficulties (see section on Disorders of Childhood Growth for details). The relationship between GH and IGF-1 in controlling postnatal growth was analyzed in mouse mutants lacking GHR, IGF-1, or both. This demonstrated that GH and IGF-1 promote postnatal growth by independent and common functions.
Thyroid Hormone
Thyroid hormone is required for normal linear growth. Newborns with hypothyroidism are of normal birth length, suggesting that fetal growth is not greatly affected by fetal thyroid hormone production. In contrast, severe, long-standing postnatal hypothyroidism can impair linear growth and delay skeletal maturation. Mutations in either thyroid hormone receptor-alpha or -beta cause some impairment in linear growth, suggesting that both of these receptors contribute to linear growth regulation. Thyroid hormone has both a direct effect on growth plate chondrocytes, particularly supporting chondrocyte hypertrophy, and an indirect effect, mediated by a stimulatory effect on growth hormone secretion.
Glucocorticoids
Glucocorticoids, when present in concentrations that exceed physiological levels, impair linear growth. This linear growth impairment arises in part from a direct effect of glucocorticoids on growth plate chondrocyte proliferation. Glucocorticoids may also indirectly affect growth plate chondrogenesis by altering GH secretion. However, not all studies show an effect, and any effects may be caused by the glucocorticoid-induced obesity. Circulating concentrations of IGF-1, a marker and mediator of GH action, do not appear to be reduced by glucocorticoid excess, which would be consistent with the idea that the decreased GH concentrations are primarily caused by obesity. The inability of GH treatment to fully compensate for the linear growth impairment caused by glucocorticoid excess further suggests that the growth impairment is primarily caused by a direct action of glucocorticoids on growth plate chondrocytes.
Estrogens
In adolescents, both male and female, estrogens stimulate linear growth, contributing to the pubertal growth spurt. In boys, the estrogen that stimulates the adolescent growth spurt is produced by aromatization of testicular and adrenal androgens. The growth acceleration is mediated in part by the stimulatory effect of estrogens on GH secretion. In addition, estrogen appears to have direct actions on growth plate chondrocytes. Estrogen stimulates growth plate senescence, the developmental program in the growth plate that is responsible for the progressive slowing of linear growth with age. Consequently, estrogen exposure leads to more rapid “aging” of the growth plate, earlier growth cessation, and earlier epiphyseal fusion. This effect of estrogen accounts for the early epiphyseal fusion in children with precocious puberty and the late epiphyseal fusion in individuals with hypogonadism, aromatase deficiency, and estrogen resistance.
Thus the effect of estrogen on linear growth is a “two-edged sword” in that it has two opposing effects on adult height. Estrogen stimulates the linear growth rate, which would be expected to increase adult height, but also accelerates growth plate senescence, which would be expected in decreased adult height. In general, the latter effect predominates. As a result, untreated precocious puberty can cause initial childhood tall stature but eventual adult short stature, whereas untreated hypogonadism or delayed puberty can cause adolescent short stature but eventual adult tall stature. These concepts also suggest that gonadotropin-releasing hormone (GnRH) analogues and aromatase inhibitors would increase adult height (see section on Treatment of Short Stature, later in this chapter).
Androgens
Androgens also can accelerate linear growth and thus contribute to the pubertal growth spurt. Androgens, secreted by the gonads or adrenal glands can be aromatized to estrogens in various peripheral tissues, including adipose tissue and thereby affect linear growth. Aromatase is also expressed in growth plate cartilage ; thus local conversion into estrogen may occur. In addition to androgen’s role as a precursor for estrogen, androgen per se appears also to stimulate linear growth. For example, dihydrotestosterone, which is not aromatizable, appears to accelerate linear growth in adolescent boys. Animal and in vitro studies suggest that these effects are mediated, at least in part, through a direct effect on the growth plate.
Insulin
Insulin positively regulates human fetal growth. Consequently, fetal growth retardation occurs in individuals with decreased fetal insulin production because of permanent neonatal diabetes mellitus and those with decreased insulin sensitivity because of severe biallelic insulin receptor defects. Conversely, increased fetal growth occurs in pregnancies complicated by gestational diabetes. This macrosomia, which primarily involves weight rather than length, is thought to be caused by the high glucose concentrations stimulating high fetal insulin concentrations.
Nutritional Regulation of Linear Growth
Inadequate nutritional intake causes functional GH insensitivity, with decreased circulating IGF-1 concentrations and increased GH concentrations. Proposed mechanisms include decreased insulin concentrations, causing downregulation of GHR expression and increased FGF-21 concentrations, causing a decrease in hepatic GHR and phosphorylated STAT5 (which is involved in GH signal transduction). The increased FGF-21 concentrations in malnutrition may impair GH sensitivity not only in the liver but also in the growth plate. In addition, undernutrition can decrease thyroid hormone concentrations, as part of the nonthyroidal illness syndrome, increase cortisol concentrations, and decrease sex steroids (mediated by decreased leptin and hence gonadotropins), all of which may contribute to the decrease in linear growth.
Conversely, overnutrition can stimulate linear growth. Obese children tend to be tall with an increased bone age. Because of the accelerated skeletal maturation and, in girls, the earlier puberty, there is little effect on adult height. Proposed mechanisms by which obesity affects linear growth include increased levels of estrogens because of peripheral aromatization and increased free IGF-1, leptin, prolactin, and adrenal androgens.
Cytokine Regulation of Linear Growth
Impaired linear growth often occurs in children with systemic inflammatory disorders, including juvenile idiopathic arthritis (JIA), IBD, and cystic fibrosis. The underlying mechanisms are likely complex and involve multiple mediators, including malnutrition and glucocorticoid excess. In addition, there is evidence that elevated levels of proinflammatory cytokines contribute to the poor growth. Various proinflammatory cytokines, including tumor necrosis factor-α (TNF-α), IL-1β, and IL-6, act directly on growth plate chondrocytes to suppress bone growth. Proinflammatory cytokines may also act indirectly on the growth plate by decreasing circulating IGF-1, sex steroid, and thyroid hormone concentrations. The effects of these cytokines may be mediated in part by upregulation of an intracellular protein called SOCS, which decreases GHR signaling.
Autocrine/Paracrine Regulation of Linear Growth
Many different autocrine/paracrine factors are secreted by growth plate chondrocytes and act locally on other chondrocytes. These factors serve to orchestrate the complex processes of proliferation and differentiation in the growth plate. Consequently, genetic defects in these local growth factors, their receptors, or other genes involved in growth factor signaling can impair growth plate chondrogenesis and therefore linear growth of the child. Depending on the specific paracrine system involved and the severity of the genetic abnormality, the bones might be both short and malformed, presenting clinically as a chondrodysplasia, or short but not malformed, presenting clinically as isolated short stature. Following are some examples of autocrine/paracrine growth factors that regulate growth plate chondrocytes and are clinically important.
C-Type Natriuretic Peptide
C-type natriuretic peptide (CNP) was named based on its structural similarity to atrial and brain natriuretic peptides. However, CNP serves different functions. It acts locally in the growth plate to stimulate chondrocyte proliferation and hypertrophy. Consequently, loss-of-function mutations in NPR2, the receptor for CNP, cause short stature whereas gain-of-function mutations cause tall stature (see section on Disorders of Childhood Growth for details). CNP analogues are in clinical trials to augment bone growth in achondroplasia (ACH).
Fibroblast Growth Factors
FGFs, acting through FGF receptor-3 (FGFR3), negatively regulate growth plate chondrogenesis. As a result, gain-of-function mutations in FGFR3 impair linear growth. Depending on the severity of the mutation, the clinical presentation can include several skeletal dysplasias with severe short stature. Recent evidence suggests that milder gain-of-function mutations can present as isolated short stature without an evident skeletal dysplasia. Conversely, loss-of-function mutations cause tall stature.
Insulin-like Growth Factors
IGF-1 and -2 act through the receptor IGF1R to stimulate growth of many tissues. In the growth plate, they act on chondrocytes to stimulate proliferation and hypertrophy. IGF-1, produced primarily in the liver, acts in an endocrine fashion on the growth plate (see section Regulation of Linear Growth-Endocrine Regulation of Linear Growth), but IGF-1 is also thought to be produced locally in the growth plate, by cells in the perichondrium and/or chondrocytes, and act locally on chondrocytes as a paracrine factor. In rodents, both endocrine and paracrine effects are important, but the relative importance in humans is unclear. IGF-2 acts primarily as a paracrine factor and is expressed at high levels in fetal tissues. The IGF2 gene is imprinted and consequently is expressed only by the paternal allele. In humans, mutations in IGF1, IGF2, and IGFR1 can all impair both fetal and postnatal growth (see section on Disorders of Childhood Growth for details) indicating that both IGF-1 and IGF-2 are important for growth before and after birth.
Parathyroid Hormone-Related Protein and Indian Hedgehog
Parathyroid hormone-related protein (PTHrP) is expressed in the chondrocytes near the ends of the long bones in the embryo and in the resting zone of the growth plate postnatally. It diffuses down through the growth plate. PTHrP acts through PTH1R (the same receptor used by PTH) and, in part, through Gs-alpha, to prevent proliferative zone chondrocytes from undergoing hypertrophic differentiation until they reach a sufficient distance from the source of PTHrP. Indian hedgehog (IHH) is produced by chondrocytes that have just begun hypertrophic differentiation. IHH stimulates production of PTHrP, forming a negative feedback loop. Mutations in genes that encode components of this system impair bone growth. For example, loss-of-function mutations in PTH1R cause Blomstrand lethal chondrodysplasia, and gain-of-function mutations in PTH1R cause Jansen metaphyseal chondrodysplasia. Similarly, loss-of-function mutations in IHH cause brachydactyly type A1 or short stature with nonspecific skeletal abnormalities, and loss-of-function mutations in Gs-alpha cause Albright hereditary osteodystrophy. Mutations in genes affecting subsequent steps in the PTHrP signal transduction pathway cause acrodysostosis.
Cartilage Matrix Regulation of Linear Growth
Growth plate chondrocytes secrete a cartilage matrix that serves a structural function, allowing the growth plate cartilage to bear the considerable mechanical forces required of the skeletal system. The cartilage matrix also provides the milieu through which endocrine and paracrine factors must travel to reach growth plate chondrocytes. This matrix is a complex mixture of molecules, including multiple types of collagen, for example, collagen types II and X, and proteoglycans, such as aggrecan. Mutations in many of the encoding genes, for example, COL2A1 , COL10A1 , and ACAN (aggrecan), cause chondrodysplasias. Heterozygous mutations in ACAN can also present as short stature with advanced bone age (see section on Disorders of Childhood Growth for details). Mutations in the collagen 1 genes, COL1A1 and COL1A2 , cause osteogenesis imperfecta. Affected patients have bone fragility, reflecting the important role of collagen type I in bone matrix, but also can have short stature.
Intracellular Regulation of Linear Growth
Proliferation and hypertrophic differentiation of growth plate chondrocytes is also under complex control by intracellular factors. For example, the short stature homeobox-containing gene ( SHOX) encodes a transcription factor that is required for normal growth plate function and consequently mutations in SHOX cause short stature. Another example of an intracellular system that regulates growth plate chondrocyte function is the RAS-mitogen-activated protein kinase (MAPK) pathway. It is a signal transduction system that regulates cell proliferation and differentiation in many tissues. Ligand binding to tyrosine kinase receptors causes signal transduction involving phosphorylation of a series of proteins. Mutations that augment RAS-MAPK signaling lead to Noonan syndrome (NS), which includes short stature.
Growth Plate Senescence
Linear growth is extremely rapid in the human fetus and progressively slows during infancy and childhood. This deceleration is briefly interrupted by the pubertal growth spurt but then the deceleration resumes, causing growth to eventually cease. The decline in linear growth is caused by a developmental program intrinsic to the growth plates termed growth plate senescence . This developmental program includes a functional decline in chondrocyte proliferation and also a gradual involution of the growth plate with a decrease in the number of chondrocytes and a decrease in the height of the growth plate. Growth plate senescence advances until chondrocyte proliferation essentially ceases. At that point, the growth plate is no longer causing bone elongation, and the now-inert cartilage is remodeled into bone, an event termed epiphyseal fusion . Thus epiphyseal fusion is not the cause, but rather the effect, of growth cessation. The bone age (usually assessed by a radiograph of the nondominant hand and wrist) serves as a radiological marker for growth plate senescence. As a consequence, the bone age provides an estimate of how far senescence has proceeded in a child and therefore how much of the child’s growth potential has been expended and how much remains. For this reason, the bone age is useful for prediction of adult height.
Growth-inhibiting conditions, such as malnutrition, hypothyroidism, and GH deficiency, generally slow not only the growth rate, but also the rate of growth plate senescence. This delay in growth plate senescence is reflected by the delay in the bone age. From an evolutionary standpoint, this delay in growth plate senescence likely serves an important function in the malnourished child, allowing growth to slow to conserve nutrients, while conserving much of the growth potential for better times.
In contrast, estrogen accelerates growth plate senescence, explaining the advanced bone age and loss of adult height in children with precocious puberty and explaining the delayed bone age and increase in adult height of adolescents with hypogonadism, aromatase deficiency, and estrogen resistance. For example, in a man with biallelic mutations in estrogen receptor-alpha, linear growth continued at a slow rate well into adulthood, resulting in tall stature, and epiphyseal fusion occurred at 35 years of age. This effect of estrogen on growth plate senescence also explains the potential therapeutic use of GnRH analogues and aromatase inhibitors to increase adult height.
Catch-Up Growth
Catch-up growth occurs in children following a period of growth inhibition. Linear growth can be inhibited by many different conditions, including malnutrition, hypothyroidism, growth hormone deficiency, and glucocorticoid excess. If the condition persists long enough, the child’s height standard deviation score (SDS) for age will decline (decreasing percentile on height curve). Often the growth-inhibiting condition will eventually resolve, for example, if food becomes more available, thyroid hormone or growth hormone replacement is initiated, or pharmacological glucocorticoids can be stopped. In this situation, the child’s linear growth rate usually does not just increase to equal the normal growth rate for age. Instead, the child’s growth rate exceeds normal, causing the height SDS to improve. This catch-up growth is often not complete in that the child’s height SDS does not return all the way back to the value before the growth inhibition, and the adult height may still be somewhat compromised.
For some time, it was thought that catch-up growth results from a mechanism within the CNS that assesses the child’s actual body size, compares this actual body size to age-appropriate set point, and then adjusts the growth rate accordingly. After a period of growth inhibition, this hypothesized mechanism would sense the height deficit and increase the growth rate to cause catch-up growth. However, more recent evidence suggests that the primary mechanism responsible for catch-up growth resides not in the CNS but rather in the growth plates and involves delayed growth plate senescence. Growth-inhibiting conditions slow growth plate senescence. When the growth-inhibiting condition resolves, the growth plates are less senescent than normal and therefore grow more rapidly than normal and for a longer period of time, causing catch-up growth. In some conditions, the rate of human catch-up growth fits this hypothesis, but in others conditions the high initial rate of catch-up growth suggests the possibility of other contributing mechanisms.
Normal Variation in Stature
Childhood growth patterns and adult height are the result of interactions between genetic and environmental factors. Since Francis Galton’s famous study in 1885, it has been known that a child’s height is strongly correlated with the height of his or her parents. Later large-scale studies focusing on height variability between twins showed that environmental factors have greater importance during early childhood, whereas genetic factors have a more decisive influence on the height of adolescents and adults. Recently, the specific genes that control normal height variation have begun to be elucidated by genome-wide association studies (GWAS), which search for statistical associations between adult height and single nucleotide polymorphisms. These studies have identified common allelic variants (usually with a minor allele frequency > 5%) that individually exert only a small phenotypic effect, but together determine much of the variability of height. This approach was used to identify variants in more than 700 loci that have a significant influence on height. Each of these variants is responsible for approximately 1 mm in height variability per allele and in combination, explain approximately 25% of the heritability in adult height. In addition, 83 lower-frequency coding variants (minor allele frequency ≤ 1%) have been identified as being responsible for up to 2 cm of height per allele.
Clinical Evaluation of Growth
Measurement
Accurate measurement is the critical cornerstone of all growth evaluations. Although it is often dismissed as simple (after all, how many parents track their kids’ growth on a wall in their homes?), two factors make it in fact challenging. First, because growth is defined as the change in length or height over a certain period of time, the two measurements at each end of that time interval must be performed according to identical technique; any deviations by one or the other measurer can introduce artifactual microaccelerations or microdecelerations into the calculated growth that become amplified errors when the velocity is annualized for analysis ( Box 11.1 ). Second, intrinsic to identical technique is correct positioning each and every time, which can be challenging with an infant or child who is fidgety, anxious, bored, developmentally unable to understand or execute the instructions, or physically challenged in achieving and maintaining the correct position (e.g., because of hypotonia, hypertonia, contractures, balance issues, leg length asymmetry, malformations, injury, or illness). Further compounding the difficulty in maintaining identical technique each and every time are both personnel issues (the same child is commonly measured by different staff members at serial visits because of the time that has lapsed or different locations of care) and equipment issues (which must be calibrated regularly and used appropriately). A final source of error can occur at the recording stage, when the measurement is entered into the medical record, such as transposing digits, other typographical error, or inadvertently recording the measurement in inches or centimeters when it had been measured in the other unit.
<SPAN role=presentation tabIndex=0 id=MathJax-Element-1-Frame class=MathJax style="POSITION: relative" data-mathml='Annualized growth velocityunits/yr=heightattime2minusheightattime1×12months#monthsbetweentimes1and2′>Annualized growth velocity(units/yr)=(heightattime2minusheightattime1)×12months#monthsbetweentimes1and2Annualized growth velocityunits/yr=heightattime2minusheightattime1×12months#monthsbetweentimes1and2
Annualized growth velocity units / yr = height at time 2 minus height at time 1 × 12 months # months between times 1 and 2
Standing height should be measured with a wall-mounted stadiometer. The child should be standing erect against the wall or backboard with shoes off, any interfering hair accessories removed, and the head in the Frankfurt plane (the lower margins of the orbits and the upper margins of the ear canals all lying in the same horizontal plane), that is, looking straight ahead. The head plate, which is lowered on top of the head for the measurement reading, must be firmly fixed such that it is perpendicular to the wall or backboard. Flip-up horizontal bars (floppy arms) on weighing scales, frequently used to measure height, are subject to great errors because of the child’s slumping posture and considerable variation in the angle of the horizontal bar.
Recumbent length is similarly measured for infants and toddlers using a recumbent length board, composed of a firm platform, a fixed head plate, and a moveable footplate for reading the measurement. Two people are required for accurate length measurement: one to hold the head in the Frankfurt plane, looking straight up, and the second to keep the knees straight and bring the measuring board to the feet, which must also be secured perpendicular at the ankles. Many practitioners, measuring infants unassisted, lie the patient on the paper covering the examining table, mark off with a pen on the paper the positions of the head and feet, remove the patient, and then use a measuring tape to quantify the distance between the two pen markings. This common technique is highly inaccurate because of incorrect positioning of the infant, movement and crumpling of the paper, and failure to get perpendicular markings by the pen.
Despite well-established, generally accepted protocols for measuring children, studies have shown that it is frequently done improperly. Observation of 55 US primary care practices (44 pediatric, 11 family practice) found measurements were considered accurate (defined as ≤ 0.5 cm) only 30% of the time, and differed from measurements of the same children by the study investigators by an average of 1.3 cm. Because the average error exceeded the difference between normal (5 cm/year) and subnormal (4 cm/year) growth velocities, measurement error likely contributes to both inappropriate investigation and/or subspecialist referrals of normally growing children and missed detection of true growth problems in others. However, this is readily corrected through training. Randomization of these same 55 practices to educational intervention and control groups found that measurement accuracy rates rose persistently in the intervention group at 3 months (55% vs. 37% in the control group) and 6 months (70% vs. 34% in the control group) later. Although many busy primary care practices measure patients only once, triplicate measurements are taken (with the average recorded) as a means of improving accuracy when precision is key (e.g., for children undergoing growth evaluations and treatment in endocrine clinics or participating in research studies).
With the proper equipment, experienced personnel, and a cooperative child, the standing height percentile can be assessed with considerable accuracy. In contrast, even with good measurement technique, the error in short-term growth velocity assessment is problematic. The relative error (absolute error divided by the measurement) is far greater for growth velocity than for height primarily because the denominator for the growth velocity (the small difference between two successive measurements) is so much smaller than the denominator for the height. For example, for a 5-year-old child with an observed height at the third percentile, the 95% confidence interval spans the second to fourth percentile but the confidence interval for a 12-month height velocity spans the eighth to 52nd percentiles. As a result, even with 1-year growth velocity, children can easily be misclassified as having a normal versus abnormal growth velocity. Trying to measure growth velocity over periods less than a full year results in a relative measurement error that is even greater. Growth velocity, measured over a year, was found to have little diagnostic validity to identify children with isolated idiopathic GH deficiency or Turner syndrome. Height velocity had to be measured over a 3-year period to achieve acceptable validity. Thus overreliance on calculated growth velocity may be misleading. However, it is important for the clinician to identify children with poor height and/or weight gain, which can indicate an important underlying disorder. As an alternative to calculated growth velocities, declines in height and/or weight percentiles on a standard growth chart can provide valuable clues to a diminished growth rate.
Growth Charts
Because normal growth follows a predictable pattern, growth charts are constructed depicting this pattern as sequential centile curves showing the distribution of selected body measurements (usually length or height, weight, and weight-for-length or body mass index [BMI]) in children of a given country. Growth is fastest during the fetal period and gradually slows during infancy, with an approximate length increase of 25 cm over the first year of life and 10 to 15 cm over the second year. During infancy, children commonly cross length centiles (physiological rechanneling) as they transition from prenatal to postnatal growth determinants; maternal and pregnancy health and nutrition have a major impact on the former, whereas the latter normally aligns with the child’s genetic height potential. After the second birthday, the linear growth rate continues to decline, but more gradually, and shifting centile channels is abnormal. With the onset of puberty, statural growth accelerates, resulting in the pubertal growth spurt, after which the decline in linear growth rate resumes, causing height gain to gradually grind to a halt. However, attainment of adult body composition continues in the transition period that follows.
The population selected for reference is important when judgments are made about the height and growth of an individual. Multiple growth charts have been constructed for US children in past decades, differing in the racial and socioeconomic composition of the reference populations upon which they were based. Growth charts for children of other countries are likewise available, reflecting the different height distributions of the local populations. The most recent and currently recommended US growth charts, published in 2000 by the Centers for Disease Control and Prevention (CDC; available at https://www.cdc.gov/growthcharts/cdc_charts.htm ), are based on several cross-sectional data sources and introduced US pediatric BMI charts. Because the CDC and other national charts are used for clinical judgment despite being reference charts (i.e., observationally describing how children in the populations grow), the World Health Organization (WHO) set out to create growth standard charts (i.e., depicting how healthy children should grow). The 2006 WHO growth charts (available at: https://www.cdc.gov/growthcharts/who_charts.htm ) are based on longitudinal measurements from birth to age 2 years of 882 children from six different countries, representing racial diversity yet growing under “ideal” conditions, and cross-sectional data on other children after age 2 years. Thus the CDC recommends use of the 2006 WHO growth charts under age 2 years and the 2000 CDC growth charts after age 2 years in assessing growth of US children. One important distinction is that the WHO charts are based on breastfed infants versus the mostly formula-fed infants of the CDC charts, resulting in differently shaped weight curves that can alter the perceived rates of failure to thrive depending on the feeding modality of the infants assessed clinically.
Two special categories of growth charts are also helpful. Velocity charts plot annual increments rather than height (distance) and are tempo adjusted, allowing for the variation in timing and intensity of the adolescent growth spurt. These charts may be helpful to assess the growth patterns of children with slower or more rapid tempo of maturation (“early and late bloomers”), whose variations are effectively lost in the cross-sectional height charts that average growth across peers in different developmental periods. One limitation to such longitudinal growth charts is that their data were derived from relatively small, nonrepresentative population samples recorded a long time ago. A second limitation is that the large relative error in growth velocity measurements, discussed earlier, creates both uncertainty in the individual child’s data and also in the reference data upon which the growth velocity charts were based. Thus based on studies of measurement error, Voss and colleagues concluded, “Although velocity charts are attractive in concept, they seem to be no more discriminating than height charts in practice, and may be clinically deceptive unless interpreted with great care.” Alternatively, growth charts that show height, rather than growth velocity, but are based on longitudinal data, may be helpful for children with slow or rapid tempo of maturation.
The second special category of growth charts includes specific charts for children with particular genetic syndromes. These charts are used in essentially the same way as the charts for normal children. Deviation of growth from the appropriate disease-related growth curve suggests the possibility of a second underlying problem, such as acquired hypothyroidism in a patient with Down or Turner syndrome.
Body Proportions
The skeleton does not grow proportionally. The upper-to-lower body segment ratio starts at 1.7 at birth, indicating a trunk considerably longer than the lower extremities. However, after birth, the lower extremities grow more rapidly than the trunk, and the ratio drops to approximately 1.0 by 10 years of age. Early cessation of linear growth, such as that which occurs in precocious puberty, results in persistently childlike proportions (short limbs compared with the trunk), whereas prolonged growth, such as occurs in hypogonadism, creates a long-limbed habitus. Limb to trunk proportions can be assessed by three different techniques, all incorporating standing height measurement for comparison. For the upper-to-lower segment ratio, the lower body segment is measured from the upper border of the pubic symphysis to the floor while the patient is standing straight. This lower segment length is subtracted from the standing height to determine the upper segment length and then divided by the lower segment length to determine the ratio, which can be compared with reference data. The second technique measures the arm span (between the tips of the middle fingers with the arms fully extended perpendicular to the torso, again with the child standing straight against the wall) for comparison with the standing height. Normally arm span is less than standing height before age 8 years, equal to height at ages 8 to 12 years, and greater than height above age 12 years. The third technique measures the sitting height, which is divided by the standing height to yield the sitting height index. The sitting height index is sensitive for detecting disproportionate growth of the trunk (vertebrae) versus the lower extremities and can be used to screen for specific causes of disproportionate short stature, such as SHOX deficiency and hypochondroplasia. Excellent reference data are available from NHANES III ( Table 11.2 ).
| Age Group (years) | Mean Age (years) | Percentiles | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | M | SD | 5th | 10th | 15th | 25th | 50th | 75th | 85th | 90th | 95th | ||
| Males | |||||||||||||
| 2.0–2.9 | 2.46 | 619 | 56.9 | 2.01 | 53.7 | 54.4 | 54.9 | 55.6 | 56.9 | 58.3 | 59.0 | 59.6 | 60.3 |
| 3.0–3.9 | 3.45 | 508 | 55.5 | 1.8 | 52.6 | 53.2 | 53.6 | 54.3 | 55.6 | 56.8 | 57.5 | 58.0 | 58.7 |
| 4.0–4.9 | 4.47 | 553 | 54.4 | 1.8 | 51.6 | 52.2 | 52.6 | 53.2 | 54.4 | 55.6 | 56.3 | 56.7 | 57.4 |
| 5.0–5.9 | 5.43 | 496 | 53.5 | 1.7 | 50.8 | 51.4 | 51.8 | 52.4 | 53.5 | 54.6 | 55.3 | 55.7 | 56.3 |
| 6.0–6.9 | 6.45 | 262 | 52.7 | 1.6 | 50.2 | 50.7 | 51.1 | 51.7 | 52.7 | 53.8 | 54.4 | 54.8 | 55.5 |
| 7.0–7.9 | 7.47 | 273 | 52.1 | 0.0 | 49.7 | 50.2 | 50.6 | 51.1 | 52.2 | 53.2 | 53.8 | 54.2 | 54.8 |
| 8.0–8.9 | 8.46 | 267 | 51.7 | 0.1 | 49.3 | 49.8 | 50.2 | 50.7 | 51.7 | 52.8 | 53.3 | 53.7 | 54.3 |
| 9.0–9.9 | 9.50 | 281 | 51.4 | 0.1 | 49.0 | 49.5 | 49.9 | 50.4 | 51.4 | 52.4 | 53.0 | 53.4 | 53.9 |
| 10.0–10.9 | 10.45 | 288 | 51.2 | 1.5 | 48.8 | 49.3 | 49.7 | 50.2 | 51.2 | 52.2 | 52.8 | 53.1 | 53.7 |
| 11.0–11.9 | 11.44 | 275 | 51.0 | 1.4 | 48.7 | 49.2 | 49.6 | 50.1 | 51.1 | 52.1 | 52.6 | 53.0 | 53.6 |
| 12.0–12.9 | 12.47 | 204 | 51.0 | 1.4 | 48.7 | 49.2 | 49.5 | 50.0 | 51.0 | 52.0 | 52.6 | 52.9 | 53.5 |
| 13.0–13.9 | 13.47 | 192 | 51.0 | 1.4 | 48.7 | 49.2 | 49.5 | 50.0 | 51.0 | 52.0 | 52.6 | 52.9 | 53.5 |
| 14.0–14.9 | 14.49 | 186 | 51.1 | 1.5 | 48.7 | 49.2 | 49.6 | 50.1 | 51.1 | 52.1 | 52.6 | 53.0 | 53.6 |
| 15.0–15.9 | 15.45 | 182 | 51.2 | 1.5 | 48.8 | 49.3 | 49.6 | 50.2 | 51.1 | 52.2 | 52.7 | 53.1 | 53.7 |
| 16.0–16.9 | 16.45 | 196 | 51.3 | 1.5 | 48.9 | 49.4 | 49.7 | 50.3 | 51.3 | 52.3 | 53.2 | 53.8 | |
| 17.0–17.9 | 17.45 | 191 | 51.4 | 1.5 | 49.0 | 49.5 | 49.8 | 50.4 | 51.4 | 52.4 | 53.0 | 53.4 | 53.9 |
| 18.0–18.9 | 18.45 | 172 | 51.6 | 1.5 | 49.1 | 49.6 | 50.0 | 50.5 | 51.5 | 52.6 | 53,1 | 53.5 | 54.1 |
| 19.0–19.9 | 19.43 | 160 | 51.7 | 1.5 | 49.2 | 49.7 | 50.1 | 50.6 | 51.7 | 52.7 | 53.3 | 53.7 | 54.3 |
| 20.0–29.9 | 24.96 | 1620 | 52.3 | 1.5 | 49.8 | 50.3 | 50.7 | 51.2 | 52.2 | 53.3 | 53.8 | 54.2 | 54.8 |
| 30.0–39.9 | 34.72 | 1452 | 52.4 | 1.6 | 49.9 | 50.4 | 50.8 | 51.3 | 52.4 | 53.4 | 54.0 | 54.4 | 55.0 |
| 40.0–49.9 | 44.35 | 1195 | 52.2 | 1.5 | 49.9 | 50.4 | 50.8 | 51.3 | 52.3 | 53.4 | 53.9 | 54.3 | 54.9 |
| 50.0–59.9 | 54.89 | 835 | 52.1 | 1.5 | 49.8 | 50.3 | 50.7 | 51.2 | 52.2 | 53.3 | 53.8 | 54.2 | 54.8 |
| 60.0–69.9 | 64.83 | 1129 | 51.9 | 1.5 | 49.7 | 50.2 | 50.6 | 51.1 | 52.1 | 53.1 | 53.6 | 54.0 | 54.5 |
| 70.0–79.9 | 74.16 | 798 | 51.7 | 1.4 | 49.5 | 50.0 | 50.4 | 50.9 | 51.8 | 52.8 | 53.3 | 53.7 | 54.2 |
| 80.0–90.9 | 84.09 | 566 | 51.5 | 1.4 | 49.3 | 49.8 | 50.1 | 50.6 | 51.5 | 52.5 | 53.0 | 53.3 | 53.9 |
| Females | |||||||||||||
| 2.0–2.9 | 2.45 | 584 | 56.9 | 1.9 | 52.9 | 53.5 | 54.0 | 54.7 | 55.9 | 57.2 | 57.9 | 58.4 | 59.1 |
| 3.0–3.9 | 3.46 | 586 | 55.3 | 1.8 | 52.1 | 52.7 | 53.2 | 53.8 | 55.0 | 56.2 | 56.9 | 57.4 | 58.1 |
| 4.0–4.9 | 4.43 | 528 | 54.1 | 1.6 | 51.5 | 52.1 | 52.5 | 53.1 | 54.3 | 55.4 | 56.1 | 56.5 | 57.2 |
| 5.0–5.9 | 5.46 | 543 | 53.2 | 1.6 | 50.9 | 51.5 | 51.9 | 52.5 | 53.6 | 54.7 | 55.4 | 55.8 | 56.4 |
| 6.0–6.9 | 6.47 | 273 | 52.5 | 1.6 | 50.4 | 51.0 | 51.4 | 52.0 | 53.1 | 54.2 | 54.8 | 55.2 | 55.8 |
| 7.0–7.9 | 7.44 | 268 | 52.1 | 1.4 | 50.1 | 50.7 | 51.0 | 51.6 | 52.7 | 53.7 | 54.3 | 54.7 | 55.3 |
| 8.0–8.9 | 8.47 | 247 | 51.8 | 1.5 | 49.8 | 50.4 | 50.7 | 51.3 | 52.3 | 53.4 | 54.0 | 54.3 | 54.9 |
| 9.0–9.9 | 9.43 | 273 | 51.7 | 1.5 | 49.6 | 50.1 | 50.5 | 51.0 | 52.1 | 53.1 | 53.7 | 54.1 | 54.7 |
| 10.0–10.9 | 10.43 | 260 | 51.6 | 1.5 | 49.5 | 50.0 | 50.3 | 50.9 | 51.9 | 52.9 | 53.5 | 53.9 | 54.4 |
| 11.0–11.9 | 11.46 | 288 | 51.6 | 1.4 | 49.4 | 49.9 | 50.2 | 50.8 | 51.8 | 52.8 | 53.3 | 53.7 | 54.3 |
| 12.0–12.9 | 12.46 | 221 | 51.7 | 1.5 | 49.3 | 49.9 | 50.2 | 50.7 | 51.7 | 52.7 | 53.3 | 53.6 | 54.2 |
| 13.0–13.9 | 13.45 | 227 | 51.8 | 1.5 | 49.3 | 49.8 | 50.2 | 50.7 | 51.7 | 52.7 | 53.2 | 53.6 | 54.2 |
| 14.0–14.9 | 14.47 | 216 | 52.0 | 1.5 | 49.4 | 49.9 | 50.2 | 50.7 | 51.7 | 52.7 | 53.3 | 53.6 | 54.2 |
| 15.0–15.9 | 15.47 | 189 | 52.1 | 1.5 | 49.4 | 50.0 | 50.3 | 50.8 | 51.8 | 52.8 | 53.3 | 53.7 | 54.2 |
| 16.0–16.9 | 16.46 | 224 | 52.2 | 1.5 | 49.5 | 50.0 | 50.4 | 50.9 | 51.9 | 52.9 | 53.4 | 53.8 | 54.3 |
| 17.0–17.9 | 17.45 | 213 | 52.4 | 1.4 | 49.6 | 50.1 | 50.5 | 51.0 | 52.0 | 53.0 | 53.5 | 53.9 | 54.4 |
| 18.0–18.9 | 18.43 | 186 | 52.5 | 1.4 | 49.8 | 50.3 | 50.6 | 51.1 | 52.1 | 53.1 | 53.6 | 54.0 | 54.6 |
| 19.0–19.9 | 19.48 | 191 | 52.6 | 1.3 | 49.9 | 50.4 | 50.8 | 51.3 | 52.3 | 53.2 | 53.8 | 54.2 | 54.7 |
| 20.0–29.9 | 24.91 | 1842 | 52.9 | 1.5 | 50.2 | 50.8 | 51.1 | 51.7 | 52.7 | 53.8 | 54.3 | 54.7 | 55.3 |
| 30.0–39.9 | 34.85 | 1837 | 52.8 | 1.5 | 50.5 | 51.0 | 51.4 | 51.9 | 52.9 | 53.9 | 54.5 | 54.9 | 55.5 |
| 40.0–49.9 | 44.28 | 1328 | 52.8 | 1.5 | 50.5 | 51.1 | 51.4 | 51.9 | 52.9 | 53.9 | 54.5 | 54.9 | 55.4 |
| 50.0–59.9 | 54.83 | 979 | 52.8 | 1.5 | 50.4 | 50.9 | 51.3 | 51.8 | 52.8 | 53.8 | 54.3 | 54.7 | 55.2 |
| 60.0–69.9 | 64.82 | 1110 | 52.3 | 1.5 | 50.1 | 50.6 | 50.9 | 51.4 | 52.4 | 53.4 | 54.0 | 54.3 | 54.9 |
| 70.0–79.9 | 74.46 | 893 | 52.0 | 1.5 | 49.5 | 50.0 | 50.4 | 50.9 | 51.9 | 52.9 | 53.5 | 53.9 | 54.4 |
| 80.0–90.9 | 84.45 | 599 | 51.1 | 1.5 | 48.8 | 49.3 | 49.7 | 50.2 | 51.2 | 52.2 | 52.8 | 53.2 | 53.8 |
Disproportions can occur not only between the limbs and trunk but also in specific limb segments, categorizing patients into specific diagnostic groups. Rhizomelia refers to disproportionate shortening of the proximal limbs (humerus and femur), mezomelia to disproportionately shortened middle portion of the limbs (radius/ulna and tibia/fibula), and acromelia to disproportionately shortened distal limbs (hands and feet). Thus every child who presents with a growth problem should be evaluated for disproportionality because this information helps to narrow the differential diagnosis, including the possibility of skeletal dysplasias.
Skeletal Maturation
Normal variation in rates of maturation among children and adolescents impacts growth prognosis, as children with more time to grow can achieve taller adult heights. An anteroposterior (AP) radiograph of the nondominant hand and wrist is used to determine the child’s bone age as a quantitative determination of somatic maturation. The bone age assesses the degree to which the skeleton has ossified. In the human embryo, the skeleton first forms out of cartilage, and then later the cartilage is remodeled into bone. This process continues throughout childhood. In the long bones, this ossification begins as a primary ossification center in the middle of the bone shaft and then spreads outward. Later, secondary ossification centers form in the epiphyses and also spread outward. However, the primary and secondary ossification centers remain separated during childhood by a thin layer of cartilage, termed the growth plate , which is responsible for bone elongation and therefore height gain. In adolescence, as growth comes to a halt, the growth plate itself is finally ossified, an event termed epiphyseal fusion , leaving cartilage only at the articular surface. Because bone is more radiopaque than cartilage or soft tissues, a radiograph of the left hand and wrist reveals how far along the ossification has proceeded in an individual child. The image is then compared with published standards (Greulich and Pyle or Tanner-Whitehouse [TW] ). There are separate sets of standards for boys and girls. If, for example, a patient’s bone age image is most similar to the standard for a 9-year-old child, then the patient is said to have a bone age of 9 years. Recently, an automated method to determine bone age has been developed.
Just like other auxological measures, bone ages have normal ranges, based on age and gender. The normal ranges define the normal variance between bone age and chronological age of a child. Discordance among skeletal centers, with maturity levels differing between bone groups, can be a normal variant. Because bone age assignment is such a subjective measure, it is prudent for pediatric endocrinologists to make their own readings rather than simply rely on radiologists’ reports. This will eliminate interreader variability between repeated studies, thereby improving the validity of longitudinally following bone age progression in a child.
The bone age mirrors the tempo of physical development of the child, particularly reflecting the progression of growth plate senescence, and indicates the remaining growth potential. Knee films can be used instead at very young ages when hand/wrist films are not yet informative because of developmentally appropriate lack of ossification. The bone age is also more closely associated than the chronological age with the timing of central puberty in some situations, for example, boys with congenital adrenal hyperplasia, familial male-limited precocious puberty, and idiopathic short stature. However, surprisingly, the bone age does not predict pubertal onset better than does chronological age in normal children.
Even before obtaining a bone age, a clinician can surmise a child’s rate of maturation by examining their teeth; the degree of dental maturation tends to correlate with skeletal maturation (i.e., delayed dentition can imply delayed bone age).
Skeletal maturation, like skeletal growth, is highly regulated by endocrine, nutritional, and genetic factors. In general, factors that positively regulate linear growth, such as thyroid hormone, GH, estrogen, and nutritional intake, also positively regulate skeletal maturation, whereas factors that negatively regulate growth, such as glucocorticoids in high concentration, also negatively regulate skeletal maturation.
Prediction of Adult Height
Adult height predictions are used to try to determine which children are growing toward a normal adult height. The “normal” range for adult height can be defined by the outer centiles demarcated on the country’s growth chart, recognizing that the centiles selected for this purpose (e.g., 5 th and 95 th or 3 rd and 97 th ) are arbitrarily chosen.
Because height is a highly hereditable trait, a more specific target can be established from the heights of the child’s biological parents. It is important to measure the parents’ heights rather than rely on self-reports, which are often inaccurate. The two parents’ heights can be combined using the formulae for gender-adjusted midparental height ( Box 11.2 ), which adjust for the 13-cm (5-inch) difference between the mean adult male and female heights, and provide the midparental height ± 2 SD (1 SD would be about 5 cm, or 2 inches) as the “normal” adult height range for their offspring. However, the adjusted midparental height assumes that the child’s height is inherited from the parents in a polygenic fashion, with multiple inherited polymorphisms contributing to the child’s height. It does not apply to monogenic conditions. If, for example, one of the two parents is short because of a heterozygous mutation in SHOX and the other parent is of normal height, the offspring will tend to be either short, like the affected parent, or of normal stature, like the unaffected parent, not halfway in between. Even for polygenic inheritance, empirical data indicate that children of short parents tend not to be as short as their parents and tall children tend not to be as tall as their parents. For example, when the midparental height is –2 SD, the children’s heights tend to be closer to –1 SD.
The simplest approach to adult height prediction is to assume that the child’s height centile will remain fairly constant until adulthood. However, this method is not accurate and may fail if the child has an advanced or delayed bone age, a condition that affects growth, or an early or late timing of puberty.
There are three popular methods for predicting adult height that take skeletal maturation (i.e., bone age) into account, and all are gender specific. The most commonly used of the three methods, the Bayley-Pinneau prediction tables from 1952, uses longitudinal data derived from healthy US children living in Cleveland to estimate the fraction of growth remaining at each bone age (by the Greulich-Pyle method ). Adult height is predicted by combining the child’s bone age with the height measured at the time of the radiographic study. The prediction tables and the prediction error are provided in the Greulich and Pyle Atlas. The Greulich and Pyle bone age (read either manually or with an automated system) can also be used in a more sophisticated mathematical prediction model (currently available online), which appears to have a slightly greater accuracy than the Greulich and Pyle tables in children with idiopathic short stature. The TW method, derived from British children, uses TW standards for bone age assessment, in which each bone in the hand and wrist is assessed individually. In addition to the bone age and measured height, this method factors in chronological age, parental heights, and in girls, the occurrence of menarche. The third method for predicting adult height is the Roche-Wainer-Thissen (RWT) method. This method takes into account the weight or nutritional status of the child and uses recumbent length instead of standing height (calculated as 1.25 cm greater than standing height). The five predictor variables in the RWT method are recumbent length, weight, bone age, chronological age, and parental heights. An additional method of height prediction has more recently been developed that uses more sophisticated mathematical methods and may show better accuracy than previous methods. It is currently available online.
Unfortunately, even using bone age, height predictions are not highly accurate. The prediction error is considerable but does decrease with age. For example, using the Bailey-Pinneau tables for girls, the prediction can err for a 9-year-old child by approximately 7.4 cm or 3 inches (2 SD of measurement error), and by approximately 3 cm or 1.2 inches for a 13-year-old child. Furthermore, these prediction methods and the associated error estimates are based on data from healthy children. There may be systematic errors in the setting of illness or in idiopathic short stature. For example, these methods overpredict adult height in untreated precocious puberty, Turner syndrome, and primordial short stature. Sources of inaccuracy in adult height prediction include the inaccuracy of the bone age estimation itself, the inability to predict pubertal onset and tempo, and the difficulty taking into account effects of disease. A small difference in bone age determination can lead to a great difference in adult height prediction, especially during the pubertal growth spurt. A computer simulation, comparison study of three methods for adult height prediction—Bayley-Pinneau, RWT, and Khamis-Roche (calculates adult height without bone age from the child’s height, weight, and midparental height, using gender and age-specific coefficients) —found poor overall agreement among the three methods (κ = 0.21 for boys and negative for girls) in predicting adult height under the 1.2 nd percentile (the threshold for the US Food and Drug Administration [FDA]-approved indication of growth hormone treatment for idiopathic short stature). It is important for both clinicians and patient-families to understand the considerable limitations of height prediction.
Inheritance of Stature
Much of the normal variability of height between individuals is caused by a polygenic influence. Multiple genetic variants, both common and rare, each having a small or moderate effect on linear growth, combine to determine the individual’s height. However, short or tall stature, especially when severe or associated with other abnormalities, can also be primarily monogenic, that is, caused by a single variant that has a strong effect on linear growth. These monogenic causes may be identified because they follow classic Mendelian inheritance patterns, usually autosomal dominant or autosomal recessive and less often X-linked. Clinically, the inheritance pattern is investigated by eliciting a careful family history of the heights of the extended family members, which can be visualized with a written pedigree. For a more detailed discussion, see section on Diagnostic Approach to the Patient with Short Stature later in this chapter. When the pedigree suggests a polygenic inheritance pattern, the clinician can calculate the adjusted midparental height, which provides an estimate of the likely adult height of the child, based on the heights of the two parents. This topic is discussed in more detail in a previous section of this chapter: Clinical Evaluation of Growth—Prediction of Adult Height
Disorders of childhood growth
Many disorders affect childhood growth, particularly decreasing growth to cause short stature. Many of the major causes are discussed later. However, the list is not exhaustive. An extensive, organized list can also be found within the International Classification of Pediatric Endocrine Diagnoses (ICPED, http://www.icped.org/ ).
Short Stature
Short stature results from decreased growth plate chondrogenesis. The decreased chondrogenesis can be caused by factors intrinsic to the growth plate (primary growth impairment) or can be secondary to factors elsewhere in the body that secondarily affect growth plate chondrocytes (secondary growth impairment) ( Fig. 11.4 ).
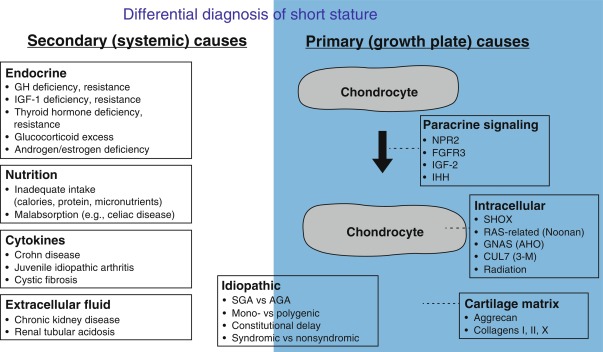
Primary growth plate impairment can involve abnormalities in paracrine signaling, cartilage matrix, or intracellular factors. In some conditions, the defect in growth plate chondrogenesis causes bones to be not only short but also malformed, in which case the condition is called a skeletal dysplasia or, more precisely, a chondrodysplasia , for example, ACH. Often, genetic defects that affect the growth plate also affect the development of other tissues, leading to a syndrome that includes not only short stature but also other congenital anomalies and organ dysfunction, for example, NS or pseudohypoparathyroidism 1A.
Secondary growth impairment can involve nutritional deficiencies, endocrine abnormalities, inflammatory disorders, and extracellular fluid abnormalities. Often these disorders are caused by a defect in some organ system other than the skeleton, such as the kidneys, pituitary, thyroid, or immune system. These disorders occurring elsewhere in the body then lead to an abnormal concentration of some molecules that are needed for normal growth plate chondrocyte function. These molecules can include steroid or polypeptide hormones, proinflammatory cytokines, or small molecules, such as phosphate or hydrogen ion.
Both primary and secondary growth impairment can be monogenic, oligogenic, polygenic, or nongenetic. Some conditions affect fetal growth, and thus the child is born small for gestational age. Other conditions affect only postnatal growth so that the child is born at a size appropriate for gestation age, and the short stature develops subsequently. Yet other conditions affect both prenatal and postnatal growth.
Causes of Short Stature
Secondary Growth Impairment
Nutritional Deficiencies
Adequate nutrition is required to support optimal statural growth, and there is often a lag between weight deceleration/acceleration and height deceleration/acceleration. Malnutrition can result from insufficient dietary intake, malabsorption or some other gastrointestinal process, or a combination of the two.
Malnutrition is the most common cause of poor growth globally, with insufficient dietary intake (total calorie and/or protein-calorie malnutrition) often related to food insecurity and poverty. Malnutrition is also common in higher socioeconomic communities, when energy demands (e.g., increased because of illness, sports or activities) are not met by intake (e.g., because of unstructured meals, picky eating, fear of obesity). Decreased nutrient intake can result from oropharyngeal malformations (e.g., Pierre Robin sequence, cleft lip/palate), abnormal oral-motor function (e.g., pervasive developmental delay), as well as loss of appetite because of certain medications (e.g., stimulants for treatment of attention deficit hyperactivity disorder or chemotherapeutic agents). Eating disorders (e.g., anorexia nervosa) involve disturbed eating behaviors related to an altered body image or food avoidance because of phobias or sensory aspects of food. Malabsorption can also cause malnutrition, and disorders such as celiac disease, IBD, or cystic fibrosis can present as isolated growth failure. Bone age and puberty are often delayed.
In addition to malnutrition, deficiency of certain micronutrients can impair statural growth. The frequent occurrence of concurrent deficiencies complicates the research and literature on any isolated micronutrient. Although iron deficiency is often found in children with nutritional growth stunting and should be corrected, multiple metaanalyses and systematic reviews of randomized controlled intervention trials of iron supplementation showed that iron therapy had no significant effect on growth in iron-replete children. In contrast, human growth retardation (and male hypogonadism) from zinc deficiency was first reported by Prasad and colleagues in 1963, and several, but not all, systematic reviews show height and/or weight increase from zinc supplementation in children. Zinc ion is required for activity of over 300 enzymes (zinc metalloenzymes), including DNA polymerase, ribonucleic acid (RNA) polymerase and thymidine kinase, which are important for nucleic acid and protein synthesis and cell division. Bone contains large amounts of zinc where it plays an important role in skeletal development, and zinc deficiency reduces IGF-1 production, cellular IGF-1 responsiveness, and growth response to GH treatment of children who are GH deficient.
Careful assessment of weight and height progression will identify children who are not gaining weight and growing appropriately. Taking a detailed dietary history is essential to establishing this diagnosis, such as a 3-day diet diary for analysis of any macro- or micronutrient deficits. Treatment is, of course, nutritional repletion, which can involve patient-family education, behavior modification, dietary supplements and in severe cases, tube feedings, as well as treatment of any underlying gastrointestinal issues. However, it is important to have in mind, that several conditions that cause short stature are associated with low BMI (or have low BMI as a component of their phenotype), such as Silver-Russell syndrome (SRS), NS, and Bloom syndrome.
Endocrine Deficiencies
Growth Hormone Deficiency/Insensitivity
Disorders of GH/IGF axis comprise a large and heterogeneous group of conditions with distinct phenotypes ( Table 11.3 ). Clinical and hormonal evaluations are usually the cornerstones of diagnoses of these disorders.
| Hormone Measurement | |||||||
|---|---|---|---|---|---|---|---|
| Gene | Inheritance | Growth Pattern | GH | IGF-1 | IGFBP-3 | Additional Features | |
| GH deficiency | multiple | AR/AD | varying severity/postnatal | ↓ to ↓↓↓ | ↓ to ↓↓↓ | ↓ to ↓↓↓ | Midline defects, deficiency of other pituitary hormones |
| Bioinactive GH | GH1 | AR/AD | varying severity/postnatal | ↑↑ | ↓↓ | ↓↓ | Growth and IGF-1 improve after exogenous GH |
| GH insensitivity (GHI) | GHR | AR > AD | severe/postnatal | Nl to ↑↑ | ↓ to ↓↓↓ | ↓ to ↓↓↓ | Growth and IGF-1 do not improve after exogenous GH. Low GHBP in 70% of cases |
| GHI associated with immune dysfunction | STAT5b | AR > AD | severe/postnatal | ↑↑ | ↓↓ | ↓↓ | GHI phenotype with immune dysfunction and elevated PRL |
| Ternary complex defects | IGFALS | AR | mild/postnatal | Nl to ↑ | ↓↓ | ↓↓↓ | Laboratory abnormality disproportionate to mild short stature, (IGFBP-3 lower than IGF1 concentrations) |
| PAPPA2 | AR | varying severity | ↑↑ | ↑↑↑ | ↑↑↑ | Mild microcephaly. Low free IGF-1 a | |
| IGF1 defects | IGF1 | AR/AD | severe/pre- and postnatal | ↑↑ | ↓↓↓↓ | Nl or ↑ | Microcephaly, sensorineural deafness, developmental delay, intellectual disability |
| IGF2 defects | IGF2 | ADp | severe/pre- and postnatal | Nl or ↑ | Nl or ↑ | Nl or ↑ | Phenotype resembling Silver Russell syndrome and low IGF-2 concentrations a |
| Bioinactive IGF-1 | IGF1 | AR | severe/pre- and postnatal | ↑↑ | ↑↑↑↑ | Nl | Microcephaly, sensorineural deafness, developmental delay, intellectual disability, insulin resistance a |
| IGF insensitivity | IGF1R | AD > AR | varying severity/pre- and postnatal | Nl to ↑↑ | Nl to ↑↑ | Nl to ↑↑ | Variable clinical findings in the dominant forms. Microcephaly, developmental delay and intellectual disability may be present |
Growth Hormone Deficiency
GH deficiency is a rare disorder with a prevalence of approximately 1 in 4000 during childhood. GH deficiency may be isolated (IGHD) or combined with other hormone deficiencies, when it is known as combined or multiple pituitary hormone deficiency (CPHD/MPHD). The condition may be congenital or acquired ( Table 11.4 ). Early diagnosis and treatment is critical to ensure optimal final height outcomes. The diagnosis of GH deficiency is based on a combination of auxology, exclusion of other pathology leading to short stature, biochemical investigation of the GH-IGF-1 axis, and imaging of the hypothalamopituitary area. More recently, a molecular diagnosis may be sought in those with “idiopathic” GH deficiency or MPHD (see Table 11.1 ); however, the majority of patients with IGH deficiency or with CPHD remain to be explained in terms of etiology. Before evaluation of the GH-IGF-1 axis, other diagnoses, such as familial short stature, hypothyroidism, Turner syndrome, celiac disease, chronic illness, such as Crohn disease or renal failure, and skeletal dysplasias should be excluded. Consensus guidelines on the diagnosis of GH deficiency in childhood were published in 2000, ( https://doi-org.easyaccess1.lib.cuhk.edu.hk/10.1210/jcem.85.11.6984 ) and suggest investigation into the following scenarios:
- 1)
Severe short stature, defined as a height more than 3 SD below the mean.
- 2)
Height more than 1.5 SD below the midparental height.
- 3)
Height more than 2 SD below the mean and a height velocity over 1 year more than 1 SD below the mean for chronological age, or a decrease in height SD of more than 0.5 over 1 year in children over the age of 2 years.
- 4)
In the absence of short stature, a height velocity more than 2 SD below the mean over 1 year or more than –1.5 SD sustained over 2 years, which may occur in GH deficiency presenting in infancy or in organic acquired GH deficiency.
- 5)
Signs indicative of an intracranial lesion.
- 6)
Signs of MPHD.
- 7)
Neonatal symptoms and signs of GH deficiency.
| 1. Congenital |
|
|
|
|
|
|
|
|
|
|
|
|
| 2. Acquired Causes |
|
|
|
|
|
|
|
|
|
|
The presentation in the neonatal period can include hypoglycemia, prolonged conjugated hyperbilirubinemia (usually with associated ACTH deficiency), and micropenis. Birth size is typically within the normal range, although there may be a reduction of approximately 10%, which occurs typically late in pregnancy. Growth velocity is reduced during the first year of life in children with severe GH deficiency with growth failure manifesting by 6 months of age, but the phenotype evolves after 1 year of age in those with milder GH deficiency. The earliest manifestations are a reduction in height velocity followed by a reduction in height SDS adjusted for mean parental height SDS. The child’s height SDS will ultimately fall below –2 SD, the time taken depending on the severity and duration of GH deficiency. A child with severe GH deficiency often has midface hypoplasia, hypotonia, a high-pitched voice, immature appearance, delayed dentition, thin sparse hair, slow nail growth, and truncal adiposity. GH deficiency is also associated with effects on cognition. In mice with GH deficiency, reduced spatial learning and memory has been reported whereas in untreated children with GH deficiency IQ, verbal comprehension and processing speed are reduced. In children and adults, the neurocognitive defects in GH deficiency are improved by GH therapy. With prompt recombinant human GH (rhGH) treatment, the outlook for final height is excellent. Other hormone deficiencies may require treatment; in particular those with structural abnormalities of the hypothalamopituitary region are more likely to evolve and develop other hormone deficiencies.
Causes of growth hormone deficiency
Congenital disorders of hypothalamic development
Hypothalamopituitary dysfunction can arise from congenital malformations of the brain or hypothalamus or of the pituitary gland. Anencephaly results in a pituitary gland that is small or abnormally formed and frequently ectopic. Holoprosencephaly, resulting from abnormal midline development of the embryonic brain, is also typically associated with hypothalamic insufficiency. The clinical spectrum of holoprosencephaly can range from cyclopia to hypertelorism, accompanied by absence of the philtrum or nasal septum and midline clefts of the palate or lip. In these situations, the classic endocrine feature is diabetes insipidus (DI), often accompanied by GH, thyroid-stimulating hormone (TSH), and ACTH deficiencies.
Debate continues as to whether the incidence of GH deficiency is increased in cases of simple clefts of the lip and/or palate alone. Clearly, children with clefts who are growing abnormally require further evaluation, and both GH deficiency and CPHDs are more frequent in this cohort. SOD is a rare, congenital, heterogeneous anomaly with a prevalence ranging from 6.3 to 10.9 per 100,000. The condition is defined by the presence of any two of three features: midline forebrain defects, such as absence of the septum pellucidum and/or corpus callosum; optic nerve hypoplasia (ONH); and hypopituitarism caused by hypothalamopituitary maldevelopment. De Morsier, in 1956, described the postmortem findings of ONH and agenesis of the septum pellucidum and coined the term SOD , also known as De Morsier syndrome . Approximately 30% of patients with SOD manifest the complete clinical triad, 62% of patients have some degree of hypopituitarism, and 60% have an absent septum pellucidum. The condition is equally prevalent in males and females. ONH may be unilateral or bilateral and may be the first presenting feature, with the later onset of endocrine dysfunction. Bilateral ONH is more common (88% as compared with 12% unilateral cases). In addition, there appears to be little correlation between the size of the optic nerve and its visual function. Neuroradiological abnormalities are present in up to 75% to 80% of patients with ONH. Pituitary hypoplasia may manifest as endocrine deficits varying from isolated GH deficiency to panhypopituitarism. There has been some suggestion that abnormalities of the septum pellucidum and hypothalamopituitary axis on neuroimaging can predict the severity of endocrine dysfunction. A decrease in growth rate because of GH deficiency is the most common feature, with hypoglycemia and polyuria and polydipsia being less common. Either sexual precocity or failure to develop in puberty may occur. Abnormal hypothalamic neuroanatomy or function and DI may be a feature. The endocrinopathy evolves with a progressive loss of endocrine function over time. The most common endocrinopathy is GH deficiency followed by TSH and ACTH deficiency. Gonadotropin secretion may be retained in the face of other pituitary hormone deficiencies. Neurological deficit is common, but not invariably so, and deficits range from global retardation to focal deficits, such as epilepsy or hemiparesis. Other neuroanatomic abnormalities include cavum septum pellucidum, cerebellar hypoplasia, schizencephaly, and aplasia of the fornix. An association between SOD and other congenital anomalies, such as digital abnormalities, is not uncommon.
The etiology of SOD has remained largely unknown until recently. Both genetic and environmental factors have been implicated in the etiology of the condition. Mutations in genes implicated in forebrain, eye, and pituitary development account for a small number of patients with SOD, and these genes include HESX1 , SOX2 , OTX2 , and TCF7L1. Environmental agents, such as viral infections, vascular or degenerative changes, and exposure to alcohol or drugs have also been implicated in the etiology of SOD. The condition presents more commonly in children born to younger mothers and clusters in geographic areas with a high frequency of teenage pregnancies. The overall frequency of genetic mutations in SOD has, however, been low suggesting that mutations in other known or unknown genes may contribute to this complex disorder.
Recently, the Sonic Hedgehog (SHH) signaling pathway has been implicated in more complex disorders of pituitary development. Mutations within SHH are associated with holoprosencephaly. Gli transcription factors are involved in hedgehog signal transduction, and heterozygous loss-of-function mutations in GLI2 have been identified in patients with holoprosencephaly. Phenotypic penetrance is variable. In several affected individuals with GLI2 mutations, pituitary gland function was abnormal, accompanied by variable craniofacial abnormalities. Other features included postaxial polydactyly, single nares, single central incisor, and partial agenesis of the corpus callosum. More recently, mutations in GLI2 have been associated with hypopituitarism in the absence of any midline defects.
SOX2 and SOX3 are members of the SOX (SRY-related high mobility group [HMG] box) family of transcription factors and are early markers of progenitor cells; their expression is downregulated as cells differentiate. Initially SOX2 mutations had been associated with bilateral anophthalmia, severe microphthalmia, learning difficulties, esophageal atresia, and genital abnormalities. However, further phenotypic characterization has revealed the presence of anterior pituitary hypoplasia, hypogonadotrophic hypogonadism, and variable GH deficiency, often with accompanying phenotypes, such as hippocampal abnormalities, corpus callosum agenesis, esophageal atresia, hypothalamic hamartoma, and sensorineural hearing loss. Although, in the majority of patients, the magnetic resonance imaging (MRI) reveals a small anterior pituitary, in occasional cases, the pituitary is enlarged and remains so for years.
Normal hypothalamopituitary development is critically dependent upon the dosage of SOX3; over- or underdosage can lead to hypopituitarism or infundibular hypoplasia. In humans, SOX3 duplications or mutations are associated with variable phenotypes, including either IGHD or panhypopituitarism. There is variable developmental delay, and MRI usually reveals a small anterior pituitary, an ectopic/undescended posterior pituitary, and dysgenesis of the corpus callosum. Deletion of SOX3 has recently been associated with a persistent craniopharyngeal canal in association with hypopituitarism.
Trauma of the brain and/or hypothalamus
Traumatic brain injury (TBI) has been recognized as a cause of acquired hypopituitarism in a number of adult studies. Data on pediatric patients are still sporadic, but there is a growing awareness that hypopituitarism after TBI is underdiagnosed with possible negative effects on growth and development. Although the pituitary gland is protected within the sella turcica, the rich vascular network of the hypothalamus and pituitary and the structure of the pituitary stalk make it vulnerable to the effects of traumatic brain injury. The hypothalamus and pituitary have a complex vascular supply consisting of long hypophyseal vessels and a rich network of portal capillaries that surround the pituitary and infundibulum. The pathophysiology of hypopituitarism related to TBI is not clearly defined, but it is thought that it is the result of direct trauma or of vascular injury resulting in ischemia and infarction. This is supported by the anatomic findings of autopsies following head trauma, which include anterior lobe necrosis, pituitary fibrosis, hemorrhage, infarction, or necrosis of the pituitary stalk.
It is of note that the peripheral layer of anterior pituitary cells, under the capsule, receive arterial blood from the capsule and not from the system of portal veins, and this may explain why these cells and those in a small area adjacent to the posterior lobe are the only surviving cells in cases of pure anterior lobe necrosis. Somatotrope cells are located in the wings of the pituitary gland, their vascular supply comes from portal vessels, and they are vulnerable to the disruption of blood supply after head injury. On the other hand, ACTH and TSH secreting cells are located in the medial portion of the pituitary and receive blood supply from portal vessels and the anterior pituitary artery. This may explain why GH deficiency is the most common deficiency seen after TBI. Hormone deficiencies may be identified in the first days to weeks posttrauma (acute phase) or may develop over time (late effect). As there is overlap between the symptoms and signs of hypopituitarism and those of neurological-psychological sequelae of TBI, it is possible that late-evolving or partial deficiencies can remain undiagnosed for years. It is not surprising, therefore that, in different studies, the time to diagnosis ranges from 1 to 40 years. It would appear that the majority of patients show a degree of pituitary hormone dysfunction in the first days after TBI (53%–76%); there is, however, wide variation in the reported hormone responses, which reflects the differences in patient selection and time of testing. All anterior pituitary hormones can be affected. Pituitary hormone deficiencies presenting in the acute phase are usually transient, but they may persist or appear and evolve over time. In adults, the incidence of permanent hypopituitarism ranges between 23% and 69% depending on the study. The GH axis is the most frequently affected (10%–33%), followed by the gonadal (8%–23%), adrenal (5%–23%), and thyroid (2%–22%) axes. The prevalence of permanent DI varies between 0% and 6%.
Until recently, there were only sporadic reports of hypopituitarism following TBI in children, but prospective studies designed to better characterize this disorder in the pediatric and adolescent population are in progress. The incidence of hypopituitarism is reported to range from 10% to 60% and, although this is lower in children as compared with adults, it is not uncommon. In general, the long-term outcome of TBI seems to be more favorable in children. Patients with hypopituitarism after head injury may have no clinical signs and symptoms suggestive of this disorder and prompt diagnosis requires a high degree of suspicion. A consensus guideline on the screening of patients post-TBI suggests that all patients who had TBI, regardless of its severity, should undergo baseline endocrine evaluation 3 and 12 months after the event or discharge from intensive treatment unit. For children and adolescents, an algorithm for endocrine assessment and follow-up has also been suggested. However, a recent study has reported that permanent hypopituitarism is uncommon after severe brain injury in young children when stringent clinical criteria for pituitary insufficiency are used. Routine testing of pituitary function, as suggested for adults, may not be necessary in children and may lead to a high number of abnormal test results.
Perinatal trauma to the brain, hypothalamus, pituitary stalk, or anterior pituitary may result in isolated GH deficiency or in multiple deficiencies of the anterior pituitary, as may child abuse. Many published series of patients with GH deficiency indicate an increased incidence of birth trauma, such as breech deliveries, extensive use of forceps, prolonged labor, or abrupt delivery. Debate continues as to whether GH deficiency is the consequence of difficult delivery or merely reflects the perinatal consequences of fetal pituitary insufficiency.
Inflammation and infiltration of the brain and/or hypothalamus
Inflammation of the brain (meningitis or encephalitis resulting from bacterial, viral, or fungal infections) may result in hypothalamic/pituitary insufficiency, with most cases being reported as isolated case reports. Despite the bias in the selection of cases, the incidence of endocrine deficiencies likely depends on the virulence of the infectious organism, the severity and localization of the disease, and the immune status of the host. In a study of 19 adult patients who were investigated 10 to 56 months following CNS infection, 21% had ACTH deficiency and 11% had gonadotropin deficiency, whereas there was no GH deficiency or DI reported. Hypopituitarism has been reported following infection by a variety of agents, including group B streptococcus, Haemophilius influenza , and Mycobacterium tuberculosis . The hypothalamus and/or pituitary may also be involved in sarcoidosis. Sarcoidosis is a multisystem granulomatous disease of unknown etiology, which clinically affects the CNS in 5% to 10% of cases. The effects of sarcoidosis on the hypothalamopituitary axis are the result of infiltration by granulomatous tissue. On MRI, the lesion may infiltrate the hypothalamus and pituitary and cause thickening of the pituitary stalk, and it usually enhances with gadolinium. The most frequently reported endocrine abnormality is DI, which is reported in 25% to 50% of patients with neurosarcoidosis. This is followed by hyperprolactinemia although anterior pituitary dysfunction with hypogonadism has also been reported. Sarcoidosis of the nervous system has a poor prognosis, but long-term remissions have been reported with high-dose intravenous pulse methylprednisolone therapy. Hormonal defects of less than 1-year duration may respond to steroid treatment, but longer standing deficits usually persist.
Hypophysitis is an inflammation of the pituitary gland that can be either primary or secondary and can result from infection, systemic disease, or irritation from adjacent lesions. This inflammatory process mimics, clinically and radiologically, tumors of the pituitary area. There are three histological types of primary hypophysitis: lymphocytic, granulomatous, and xanthomatous. Lymphocytic hypophysitis is the most common type; it involves the anterior pituitary and may infiltrate the infundibulum and posterior lobe. Lymphocytic hypophysitis occurs mainly in young women and is associated with pregnancy or the presence of autoimmune diseases, including Hashimoto thyroiditis, Graves disease, type I diabetes, and systemic lupus erythematosus. There are rare case reports of hypophysitis in children and adolescents, and, in most cases, diagnosis has been made only after biopsy and histological examination, although in clinical practice, this is rarely performed. In many cases, hypophysitis presented with DI and hypopituitarism and preceded the diagnosis of an intracranial tumor, such as germinoma. In other cases, it can present with DI and hypogonadotropic hypogonadism. Once the diagnosis is established, management is generally conservative, unless there are signs of increased intracranial pressure or optic nerve compression.
Langerhans cell histiocytosis (LCH) is characterised by clonal proliferation and accumulation of abnormal dendritic cells that can affect either a single site or many systems causing multiorgan dysfunction. In children, the median age of diagnosis ranges between 1.8 and 3.4 years. LCH infiltrates the hypothalamopituitary area in 15% to 35% of patients with subsequent development of at least one pituitary hormone deficiency. In a multicenter French national study of 589 pediatric patients with LCH, 145 patients (25%) had pituitary dysfunction. In 60 patients, pituitary involvement was already present at the time of diagnosis and in 20 of them it was the first manifestation of the disease. Patients at high risk of pituitary involvement seem to be those with multisystem disease involving skull and facial bones, mastoid, sinuses, and mucous membranes (i.e., gums, ear, nose, and throat region). Furthermore, compared with patients without pituitary involvement, patients with pituitary involvement have a higher rate of relapse (10% at 5 years vs. 4.8% at 5 years), and a higher incidence of neurodegenerative LCH.
DI is the most frequently reported permanent consequence of LCH and the most common endocrinopathy; almost all patients with pituitary involvement have DI, which usually presents early in the course of the disease, within the first 3 to 5 years, and occasionally may precede the diagnosis of LCH. Children with LCH and DI may also have anterior pituitary hormone deficiencies, with most deficits developing in the 6 years after the diagnosis of DI. The second most common endocrinopathy is GH deficiency, which occurs in 14% of all patients with LCH and in more than 40% of patients who have pituitary involvement. In the vast majority of patients, GH deficiency is associated with DI, with a median interval of 2.9 to 3.5 years between the diagnosis of DI and development of GH deficiency. Isolated GH deficiency, or the association of GH deficiency with other anterior pituitary hormone deficiencies, occurs less commonly.
Pituitary MRI findings in patients with LCH include thickening of the pituitary stalk, suggestive of the infiltrative process, enhancing changes in the pituitary gland and hypothalamus, and absence of the bright signal of the posterior pituitary in T1-weighted images, caused by the loss of the phospholipid-rich antidiuretic hormone secretory granules. The latter is an invariable feature of patients who develop DI. Although, at the time of diagnosis of DI, 75% show a thickened pituitary stalk, only 24% have persistent stalk thickening after 5 years. These changes are variable and do not correlate with treatment or with clinical recovery as DI persists in all cases. The role of MRI in predicting the development of anterior hormone deficiencies is uncertain. It has been reported that patients who will become GH deficient are more likely to have a smaller anterior pituitary, while the size of the stalk and posterior pituitary are not significantly different.
Long-term follow-up of patients with LCH has shown that the already established hormone deficiencies cannot be reversed by treatment.
Tumors of the brain and/or hypothalamus
Tumors of the CNS are a major cause of hypothalamic insufficiency. This is especially true for midline brain tumors, such as germinomas, meningiomas, gliomas, colloid cysts of the third ventricle, ependymomas, and gliomas of the optic nerve. Although metastasis from extracranial carcinomas is rarely found in children, hypothalamic insufficiency may result from local extension of craniopharyngeal carcinoma or Hodgkin disease of the nasopharynx. Pituitary adenomas represent less than 3% of supratentorial tumors in childhood and about 3.5% to 6% of all surgically treated pediatric pituitary tumors. In most cases, they are hormonally active, arising from any of the five cell types of the anterior pituitary, and may produce prolactin (prolactinomas, 52%), ACTH leading to Cushing disease (corticotropinomas 33.3%), GH (somatotropinomas 8%), or more rarely TSH (thyrotropinomas). Nonfunctioning pituitary adenomas are rare in children (2.7%) compared with adults where they represent almost 20% of pituitary adenomas. Although the majority of childhood pituitary adenomas are prolactinomas presenting in adolescence, corticotropinomas are the most common tumors in prepubertal children. Pituitary adenomas occur in isolation or may be part of a genetic syndrome, such as multiple endocrine neoplasia type 1 (MEN1), McCune-Albright syndrome (MAS), or Carney Complex. Their pathogenesis is not clear but there is increasing evidence that dysregulation in hormone receptor signaling, changes in molecules that regulate cell cycle or are important for adhesion to extracellular matrix, as well as changes in growth factors, may be implicated. Their clinical presentation results from pituitary hormone hypersecretion or deficiencies, disruption of growth and sexual maturation, and pressure effects. On MRI, pituitary adenomas show slow uptake of gadolinium and appear as hypoenhancing lesions that may displace the pituitary stalk.
Optic gliomas
Tumors of the optic pathway represent 4% to 6% of all pediatric intracranial tumors and among these the most common are optic gliomas (65%). Most optic gliomas are low-grade lesions with favorable prognosis if treated optimally. Gliomas confined to the optic nerve have a predilection for females (60%–70%) and are associated with neurofibromatosis type 1 (NF-1) in more than half of cases, whereas 38% are sporadic. Children with sporadic gliomas were more likely to manifest increased intracranial pressure, decreased visual activity, and more commonly documented endocrine complications. The most frequent symptoms at presentation are visual defects (diminished vision, optic atrophy, strabismus, nystagmus, proptosis), ataxia, and precocious puberty. Because of their close anatomic relation to the hypothalamus and pituitary, dysregulation of the hypothalamopituitary axis is common and is caused either by the tumor itself or secondary to treatment. Premature sexual maturation is a frequently presenting symptom whereas the most common defect following cranial irradiation is GH deficiency. NF-1–associated optic gliomas can also cause GH excess and excessive growth.
Cystic lesions
Cystic lesions in the pituitary area include Rathke’s cleft cysts, arachnoidal cysts, and cystic adenomas and craniopharyngiomas. Rathke’s cleft cysts are benign cystic remnants of Rathke’s pouch. They are usually small (< 5 mm), asymptomatic, and are found in almost 20% of routine autopsies. They consist of well-differentiated columnar or cuboidal epithelial cells and the content of the cyst varies. Rathke’s cleft cysts can grow gradually and become symptomatic, especially if they have suprasellar extension. Symptoms include headache, visual defects, and pituitary dysfunction ranging from increased prolactin to pituitary hormone deficiencies. Differential diagnosis from other cystic lesions in the area is not always easy, as on MRI the cyst fluid shows variable signal intensities and therefore cysts can appear as hypo- or hyperdense. Almost 50% of Rathke’s cleft cysts show rim enhancement. Recurrence of the cyst after treatment is rare (2 of 14 patients in one series) and it is recommended that treatment should include both fluid drainage and cyst wall removal to avoid relapse.
Arachnoid cysts consist of a collection of cerebrospinal fluid (CSF)-like fluid surrounded by a wall made of arachnoid structures. They are mainly suprasellar, with only rare cases being intrasellar. Suprasellar cysts are usually diagnosed following nonendocrine symptoms, such as neurological deficits, macrocephaly, and visual symptoms. Because of the proximity of the lesion to the hypothalamopituitary area, arachnoid cysts may cause central precocious puberty, amenorrhea, and hyperprolactinemia, in addition to thyrotropin, ACTH, or GH deficiency.
Craniopharyngiomas may result in hypothalamic dysfunction (discussed later).
Irradiation of the brain and/or hypothalamus
Cranial irradiation represents an important cause of hypothalamic/pituitary dysfunction. Irradiation may directly impair hypothalamic and pituitary function, and it is not always easy to discriminate between damage at each of these levels. In addition, thyroidal and gonadal function may also be directly affected if total body irradiation or craniospinal irradiation is administered. The degree of pituitary impairment is related to the dose of radiation received. Low doses typically result in isolated GH deficiency. With higher doses, multiple pituitary hormone deficiencies are observed. Two to 5 years after irradiation, 100% of children receiving more than 3000 cGy over 3 weeks to the hypothalamic-pituitary axis showed subnormal GH responses to insulin-provocative tests.
The degree of pituitary deficiency observed increases with time since irradiation. Children who test normally at 1-year postirradiation may still develop pituitary deficiencies at later times. Even when serum GH concentrations after provocative testing are normal, measures of spontaneous GH secretion may indicate an impairment. As little as 1800 cGy has been shown to affect spontaneous GH secretion in pubertal children.
Decreased GH secretion may be further complicated by the impact of irradiation on spinal growth, which can result in short stature and skeletal disproportion. Surprisingly, cranial irradiation can also result in precocious puberty—with the consequence that epiphyseal fusion occurs at an earlier age than is ideal from the perspective of maximizing growth. GnRH analogues may be used to delay pubertal progression. The endocrinologist observing the child who has received craniospinal irradiation must consider these three factors: evolving hypopituitarism, decreased spinal growth potential, and early puberty and premature epiphyseal fusion. Care must be taken to maximize the growth potential of the patient without causing skeletal disproportion and without delaying puberty excessively or allowing growth to terminate too early.
Genetic abnormalities resulting in combined pituitary hormone deficiency
The molecular basis of SOD and other midline conditions has been previously discussed.
Recessive mutations in LHX3 have been identified in several consanguineous families, in addition to a single patient who was found to be compound heterozygous for two missense mutations within the gene. These patients usually present with multiple anterior pituitary hormone deficits, including ACTH deficiency, which was initially thought to be spared. Pituitary morphology is variable between patients with LHX3 mutations: most patients have a hypoplastic anterior pituitary with a normal posterior pituitary and midline structures conversely, an enlarged anterior pituitary has also been reported in a patient that was not evident in a previous MRI scan performed 10 years earlier. In addition, a patient with a hypointense lesion in the anterior pituitary consistent with a microadenoma has also been described. The majority of patients with LHX3 mutations reported to date have also exhibited a short rigid cervical spine with limited neck rotation and trunk movement. Again, the skeletal phenotypes can vary, and a single patient with normal neck rotation and no other syndromic features has been reported. In addition, most patients with LHX3 mutations manifest sensorineural deafness, the severity of which is also highly variable and can range from profound to very mild and may be missed in some cases. A direct role may be implicated here because LHX3 is expressed in specific regions of the inner ear in a pattern highly conserved between humans and mice, and it is likely to have a role in cochlea hair cell development.
Heterozygous mutations in LHX4 have also been reported, with all patients exhibiting GH deficiency and associated short stature on presentation, again with variable additional endocrine deficits and extrapituitary abnormalities. Initially, a heterozygous intronic mutation that abolishes normal splicing of LHX4 was reported by Machinis and colleagues in a three-generation family segregating in a dominant and fully penetrant manner. The probands presented with short stature and were found to be GH, TSH, and ACTH deficient, with anterior pituitary hypoplasia, an undescended posterior pituitary, and absent pituitary stalk on MRI. Other affected family members presented with short stature associated with IGHD and a normal posterior pituitary. Additional manifestations included a poorly formed sella turcica and pointed cerebellar tonsils. Several mutations have been subsequently described and are clearly variable in terms of penetrance and phenotype. Recently, a novel homozygous mutation in LHX4 was associated with a severe recessive phenotype leading to a lethal form of hypopituitarism, suggesting that the variable penetrance observed in the majority of heterozygous mutations may indicate the presence of a second genetic abnormality that has not been detected to date.
The gene encoding PROP1 is involved in the early determination and differentiation of multiple anterior pituitary cell lineages. Abnormalities of PROP1 result in CPHD, characterized by variable degrees of deficiency of GH, prolactin, TSH, follicle-stimulating hormone (FSH), luteinizing hormone (LH), and occasionally ACTH. The first mutations in PROP1 were identified in human patients with hypopituitarism characterized by GH, TSH, and prolactin (PRL) deficiencies in addition to reduced gonadotrophins and failure to enter spontaneous puberty. Subsequently, PROP1 mutations have been identified in patients from over 21 different countries, implicating PROP1 mutations as the most common genetic cause of CPHD accounting for approximately 50% of familial cases, although the incidence in sporadic cases is much lower. Affected individuals exhibit recessive inheritance, and the majority of mutations identified involve the DNA binding homeodomain. The mutations in PROP1 identified to date include nonsense, missense, frameshift, intronic, and deletion mutations. The majority of the mutations are predicted to result in complete loss of function by ablating DNA binding and transcriptional activation, although some missense mutations retain partial activity. By far the most common mutation, accounting for 50% to 72% of all familial PROP1 mutations in multiple unrelated families, is a 2-bp deletion within exon 2 resulting in a frameshift (often referred to as p.S109X ). This mutation likely represents a mutational hot spot within the gene, rather than a common founder mutation, and combined with the c.150delA mutation, accounts for more than 90% of all known PROP1 mutations.
Homozygosity for mutations in PROP1 is typically associated with a deficit of GH, TSH, PRL, and gonadotrophins, although the time of onset and severity of hormone deficiency varies. Most patients present with early-onset GH deficiency and growth retardation; however, normal growth in early childhood has been reported in a patient who attained normal final height without GH replacement therapy. Patients may present with gonadotropin deficiency with the evolution of other hormone deficiencies later in life. TSH deficiency is also highly variable and has been reported as the initial presenting symptom in some cases, whereas other patients show delayed onset. The frequency of ACTH deficiency increases with age. Most patients exhibit normal ACTH and cortisol concentrations in early life; however, patients as young as 6 years have been described with cortisol deficiency, emphasizing the necessity for complete and continuing clinical assessment of patients with PROP1 mutations. The spectrum of human gonadotropin deficiency is extremely variable in patients with PROP1 mutations, ranging from early hypogonadism with a micropenis and undescended testes and complete lack of pubertal development to spontaneous, albeit often delayed, onset of puberty with subsequent deficiency of gonadotropins, requiring hormone replacement therapy.
The pituitary morphology in patients with PROP1 mutations is unpredictable; most cases show a hypoplastic or normal-sized anterior pituitary gland on imaging, with a normal pituitary stalk and posterior lobe, although some reports have documented an enlarged anterior gland. Longitudinal analyses of anterior pituitary size over time have revealed that several patients with an enlarged anterior gland at initial scanning in childhood show spontaneous regression and involution, so that MRI in older patients often demonstrates anterior pituitary hypoplasia, although the size of the pituitary can wax and wane during this time. The pituitary enlargement consists of a mass lesion interposed between the anterior and posterior lobes, possibly originating from the intermediate lobe or Rathke’s pouch remnant in the cleft, although the underlying mechanism for the mass remains unknown.
The evolving nature of hormone insufficiencies in patients with PROP1 mutations suggests a progressive decline in the anterior pituitary axis, so such patients require regular monitoring for the development of hormone deficits that may not be apparent on initial presentation. The highly variable nature of the phenotype associated with PROP1 mutations, even between siblings within the same family carrying identical mutations, again implicates unidentified genetic modifiers playing a role in the severity and onset of disease pathogenesis.
POU1F1 is the human homologue of the mouse gene Pit1 and encodes a transcription factor involved in the activation of GH and PRL genes, regulation of the TSH-b promoter, and specification, proliferation, and survival of the corresponding cell lineages. Mutations within POU1F1 (PIT1) (reviewed in ) are generally associated with GH, PRL, and TSH deficiencies with variable anterior pituitary hypoplasia. Deficiencies of GH and PRL are generally complete and present early in life, whereas TSH deficiency can be highly variable. The majority of cases present with early TSH deficiency; however, in some cases, hypothyroidism occurs later in childhood. MRI of patients with POU1F1 mutations demonstrates a small or normal-sized anterior pituitary gland with a normal posterior pituitary and infundibulum, but with no extrapituitary abnormalities. More than 28 different mutations in POU1F1 have been described to date; the majority of these show recessive inheritance, including a complete gene deletion, as well as splice site mutations, whereas a small number are dominant mutations. Of these, the amino acid substitution p.R271W is the most frequent, having been identified in several unrelated patients from a variety of different ethnic backgrounds. The p.R271W substitution results in production of a protein that remains capable of binding to DNA but acts as a dominant negative inhibitor of transcription.
Studies of POU1F1 in CPHD patient cohorts suggest that the incidence of mutations in cases of sporadic CPHD is quite low (approximately 3%–6%), whereas the incidence among familial patients with hypopituitarism is much greater (25%). Overall, screening studies suggest that abnormalities of POU1F1 are a less common cause of CPHD than are abnormalities of the PROP1 gene.
Haploinsufficiency of the homeobox gene RIEG results in Rieger syndrome, an autosomal-dominant disorder that involves abnormal development of the anterior chamber of the eye, teeth, and umbilicus—with an occasional association with GH deficiency.
Genetic abnormalities of GH production and/or secretion resulting in isolated GH deficiency
It has been reported that up to 30% of patients with IGH deficiency have an affected parent or sibling. In addition to combined pituitary hormone deficiency, such as that caused by abnormalities of PROP1 and POU1F1 described previously, four Mendelian forms of IGH deficiency have been reported.
Isolated GHDIA results from deletions (6.7- 7.0- 7.6-, and 45-kb deletions have been reported) or mutations of the GH1 gene that totally block GH synthesis or secretion. Transmission of isolated GHDIA is autosomal recessive, and patients have profound congenital GH deficiency. Because GH has never been produced by the patient, even in fetal life, patients are immunologically intolerant of GH and typically develop anti-GH antibodies when treated with pituitary-derived or recombinant DNA–derived GH, although development of growth-attenuating antibodies appears to be less frequent with the newer synthetic GH preparations.
When antibodies prevent a patient from responding to GH, recombinant (rh)IGF-1 therapy should be considered. The less severe form of autosomal-recessive GH deficiency (isolated GHDIB) is likely to also be the result of mutations or rearrangements of GH1 , presumably resulting in a GH molecule that retains some function but is perhaps unstable. In addition, mutations in GHRHR also lead to GHD1B. To date, however, most patients with presumed isolated GHDIB have not demonstrated an alteration of GH1 and GHRHR and the cause of their GH deficiency remains unclear. These patients generally respond well to GH therapy, and development of clinically significant anti-GH antibodies is unusual.
Isolated GHDII is transmitted as an autosomal dominant. Such patients usually have abnormalities of GH1 that function in a dominant negative manner. The most common causes of this disorder appear to be splice site and intronic mutations that inactivate the 5’-splice donor site of intron 3, resulting in skipping of exon 3 with generation of the 17.5-kDa isoform of GH that exerts a dominant-negative effect on the secretion of the 22-kDa molecule. Transgenic mice that overexpress the 17.5-kDa isoform have a defect in the maturation of the GH-containing secretory vesicles and display anterior pituitary hypoplasia with the development of other hormone deficiencies. The 17.5-kDa isoform is retained in the endoplasmic reticulum, misfolded, and disrupts the secretory pathway. Similar mutations identified in humans are also associated with GHDII and an evolving endocrinopathy.
Missense mutations that affect GH secretion and/or action can also cause autosomal dominant IDH deficiency. Patients with the p.Arg183His mutation have impaired release of GH as, at the cellular level, the secretory granules that contain the mutant Arg183His protein are not exocytosed as effectively as those containing the wild-type hormone. Other mutations (for example, p.Pro89leu) can cause more profound and early disturbances in the secretory pathway by alteration of the orientation of the GH helices and an effect on the correct folding of the molecule.
Patients with autosomal dominant IGH deficiency show considerable variation in the severity of GH deficiency. They present with low but detectable serum GH concentrations, variable height deficit, and can show anterior pituitary hypoplasia on MRI (38%–50%). Data on pedigrees with the Arg183His or e3 + 1G > A mutations highlight the fact that patients with the same mutation can vary considerably in height (≤–4 SDS to normal) and even attain normal adult height without treatment. Patients with splice site mutations are thought to be more severely affected than those with missense mutations ; however, patients with the IvS3 + 1 or IvS3 + 2 splice site or the p.Pro89leu mutations can develop additional pituitary hormone deficiencies, including ACTH, prolactin, TSH, or gonadotropin deficiency. This evolving phenotype is unpredictable and dictates the need for lifelong follow-up of affected individuals. Another intriguing observation is that, even in patients with a genetic cause (for example, p.Glu32Ala), GH deficiency seems to reverse when they are retested at the end of growth, in the transition period before transfer from pediatric to adult services. However, this effect is temporary, and is observed in patients tested at the time of transition, and who should not be discharged from follow-up.
No convincing cases of mutations of the GHRH gene in humans have been identified. Abnormalities of the gene for the GHRHR, on the other hand, have been found in a number of pedigrees. GHRHR is a seven transmembrane domain receptor. Its expression requires the presence of the POU1F1. Homozygous or compound heterozygous mutations have been reported in GHRHR (missense, nonsense, splice site, deletions, or regulatory mutations) leading to type IB IGH deficiency. GHRHR mutations are identified in about 10% of patients with familial recessive IGH deficiency, and in about 3% of a selected cohort of patients IGH deficiency. Mutations are more likely to be identified in consanguineous pedigrees or patients from certain ethnic backgrounds, such as from the Indian subcontinent and Brazil.
Children with mutations in GHRHR have severe GH deficiency and short stature, but midfacial hypoplasia, neonatal hypoglycemia, and microphallus, as found in those with recessive GH1 mutations, are rare. The anterior pituitary may rarely be normal; in the majority it is hypoplastic. The posterior pituitary and the pituitary stalk are normal. Partial loss of function mutations may be associated with a relatively mild GH deficiency phenotype. Overall, mutations in genes regulating GH secretion are rarely identified, suggesting that novel genes remain to be identified.
Isolated GHDIII is transmitted in an X-linked manner. Little is known about the etiology of this condition, although rare mutations in SOX3 can be associated with IGHD with or without learning difficulties.
Congenital abnormalities of the pituitary
A number of reports have described the association of “idiopathic” GH deficiency with an ectopic neurohypophysis. MRI findings have been described in several series of patients with idiopathic GH deficiency. Abrahams and colleagues studied 35 patients with idiopathic GH deficiency and found that those with MRI findings could be divided into two groups: 43% had an ectopic neurohypophysis (neurohypophysis located near the median eminence), absent infundibulum, and an absence of the normal posterior pituitary bright spot, and 43% had a small anterior pituitary as an isolated finding or combined with an ectopic neurohypophysis. All in all, an ectopic neurohypophysis was found in 87% of cases with multiple pituitary hormone deficiencies but in only 10% of cases with IGH deficiency.
In other studies, however, high-resolution MRI findings of one or more of the following have been suggested to be sensitive and/or specific indicators of hypopituitarism: a small anterior pituitary, attenuated pituitary stalk, and ectopic posterior pituitary. In one study, pituitary abnormalities were found in 80% of patients with IGH deficiency and 93% with MPHD. In patients whose peak GH concentration was less than 3 ng/mL, 90% had abnormal MRI findings—compared with 39% of those with GH concentrations 3 ng/mL or more. In another study, the stalk was abnormal in 90% of patients with IGHD and was absent in 96% of patients with MPHD. Thus MRI abnormalities are common in children with IGH deficiency and MPHD and are closely associated with the severity of GH deficiency. Patients with structural abnormalities will need lifelong follow-up in adulthood owing to the risk of developing other pituitary hormone deficiencies. Patients with an ectopic posterior pituitary, however, can also show reversal of their GH deficiency on retesting at the time of transition from pediatric to adult services or in adulthood.
Tumors involving the pituitary
Many of the tumors that affect hypothalamic function also directly impact pituitary secretion of GH. In particular, craniopharyngiomas comprise a major cause of pituitary insufficiency. These tumors arise from remnants of Rathke’s pouch, the diverticulum of the roof of the embryonic oral cavity that normally gives rise to the anterior pituitary. Some consider this tumor a congenital malformation because it is believed to be present at birth, gradually growing over the ensuing years and decades. The tumor arises from rests of squamous cells at the junction of the adenohypophysis and neurohypophysis. As it enlarges, it forms a cyst that contains degenerated cells and it may calcify but does not undergo malignant degeneration. In children, they represent 5% to 15% of intracranial tumors and are the most common neoplasm of the hypothalamopituitary area, accounting for up to 80% of tumors in this location. Their incidence in the United States is 1.3 per million per year and almost 28% affect children younger than 14 years. There is a bimodal peak in incidence: the first peak occurs in children between 5 and 14 years and the second peak in adults older than 50 years. They can, however, be diagnosed at any age and have even been reported in the neonatal period.
At presentation, most craniopharyngiomas have a combined intra- and suprasellar location (74.2%) and almost half have hypothalamic involvement (51.6%). A smaller percentage is exclusively suprasellar (22.6%) or confined within the sella turcica (6%–3%). Almost one-third invade the floor of the third ventricle and may cause obstructive hydrocephalus. In pediatric patients, craniopharyngiomas are predominantly cystic (56.7%), multicystic (16.7%), predominantly solid (13.3%), purely solid (10%), or purely cystic (3.3%). The cystic fluid is viscous and rich in cholesterol and the incidence of calcification is much higher in children (83.3%) compared with adults.
There are two main histological types: the most common is the adamantinomatous type and consists of epithelial neoplastic cells that resemble those found in lesions of the jaw. The papillary type is much rarer and is found almost exclusively in adults. Although craniopharyngiomas are histologically benign, they can extend from their initial site, develop papillae, and invade vital surrounding tissues, including the hypothalamus and optic chiasm. This attachment makes their complete excision difficult, if not impossible, and contributes to tumor recurrence and morbidity.
Craniopharyngiomas are typically sporadic tumors. There have been, however, rare case reports of affected family members suggesting recessive inheritance. Overactivation of the Wnt signaling pathway appears to play a role in craniopharyngioma development; activating mutations in beta-catenin, a downstream component of the Wnt signaling pathway, have been identified in adamantinomatous craniopharyngiomas.
Clinical signs and symptoms of craniopharyngiomas can arise at any age, from infancy to adulthood, but most typically in midchildhood. The most common presentation is with symptoms of increased intracranial pressure (up to 75%), such as headaches, vomiting, or oculomotor abnormalities. Impaired vision is common. Visual field defects may result from compression of the optic chiasm, and papilledema or optic atrophy may be observed. Visual and olfactory hallucinations have been reported, as have seizures and dementia.
It is estimated that 70% to 80% of children have evidence of endocrine deficiencies at presentation and that growth failure is observed in 32% to 52% of children before diagnosis. Low concentrations of IGF-1 have been reported in 80% of children at time of diagnosis. GH deficiency is the most common hormone deficiency, documented in 75% to 100% of those tested before treatment. It is followed by ACTH (20%–70%) and TSH (3%–30%) deficiencies. Compression of the pituitary stalk or damage to hypothalamic dopaminergic neurons results in elevated PRL concentrations, observed in 8% to 20% of children at diagnosis. The incidence of DI at presentation varies between 10% and 29% of patients, depending on the study. In adolescence in particular, craniopharyngiomas may present with delayed puberty or pubertal arrest. Rare presentations include precocious puberty and syndrome of inappropriate antidiuretic hormone secretion.
Lateral skull films often demonstrate enlargement or distortion of the sella turcica, frequently accompanied by suprasellar calcification(s). Nevertheless, some children with craniopharyngiomas will have normal plain films, and alternative radiological techniques are recommended. Computed tomography (CT) is a sensitive technique for identification of small amounts of calcification or cystic abnormalities. MRI is probably the most sensitive technique. Craniopharyngiomas appear as mass lesions in the sellar and/or suprasellar area that may extend to the hypothalamus and invade the third ventricle. Adamantinomatous craniopharyngiomas are predominantly cystic and the cystic portion of the lesion appears hyperintense in T1 and T2 images. The solid part of the tumor shows areas of high and low signal intensity that represent areas of calcification and hemosiderin deposits. In the majority of cases (58%–76%), the size of craniopharyngiomas, as estimated by MRI or CT, has been reported to be 2 to 4 cm, whereas it is smaller than 2 cm in 4% to 28% cases and more than 4 cm in 14% to 20%.
The management of craniopharyngiomas is complex, controversial, and it is best achieved by a multidisciplinary approach. Aims of the treatment are to relieve acute signs and symptoms of compression (raised intracranial pressure, threatening visual failure); to preserve hypothalamic function, thus reducing later morbidity and mortality; and to provide long-term control and prevent recurrence. What has been evident from different studies is that the extent of surgical resection is probably the most important factor that influences the recurrence of craniopharyngioma. In patients who had surgery only, the 10-year recurrence free survival rate was 83% after total removal, 50.5% after subtotal removal and 15.6% after partial removal. Tumor recurrence usually occurs in the first 5 years and is relatively rare thereafter. However, even after complete resection confirmed radiologically, relapses occur in up to 15% to 25% of patients. In older series, however, when patients were treated with radical and repeated surgical resections, mortality was high (25%–50%) and hypothalamic, visual, and cognitive morbidity occurred in the vast majority (75%), especially in craniopharyngiomas with suprasellar or retrochiasmatic extension.
Guidelines for the multidisciplinary management of children with craniopharyngioma have recently been published. It is now suggested that patients can be categorized into two risk groups with respect to management and prognosis. The good risk group includes older children with small tumors (2–4 cm) and no hypothalamic syndrome or hydrocephalus. Younger children, with larger tumors (> 2–4 cm) and hypothalamic syndrome or hydrocephalus are in the poor risk group. Complete radical resection, with or without adjuvant radiotherapy, is suggested for the good risk group, whereas limited surgery and immediate or delayed radiotherapy is the treatment of choice for the poor risk group. The recent availability of proton beam therapy is a valuable addition to the therapeutic armamentarium.
Idiopathic pituitary growth hormone deficiency
As discussed previously, many of the disease processes that affect hypothalamic regulation of GH secretion also impact pituitary function. Given current diagnostic limitations, it is not always possible to completely discriminate between hypothalamic and pituitary dysfunction. Furthermore, it is likely that many cases of so-called idiopathic GH deficiency will be found to have a molecular basis for the disorder. Indeed, in the past 5 years we have seen an explosion of information concerning genes critically involved in pituitary somatotrope differentiation and function.
At this time, however, a clear cause for pituitary GH deficiency is often not identified—hence the term idiopathic . An incidence of pituitary GH deficiency in 1:60,000 live births was reported from the United Kingdom. A more recent survey of 48,000 Scottish schoolchildren has indicated an incidence as high as 1:4000, whereas the best estimate available in the United States population is at least 1:3480.
It is likely, however, that childhood GH deficiency is an overdiagnosed condition. In particular, the diagnosis of later onset idiopathic IGH deficiency should always be questioned. Although one may argue that destructive or inflammatory lesions of the hypothalamus or pituitary may only affect GH secretion, that IGH deficiency caused by a mild mutation/deletion of the GHRHR gene or GH1 gene may appear late, or that CPHD may first present with what appears to be IGH deficiency, such circumstances appear to be rare. In the absence of anatomic abnormalities evident on imaging studies and/or biochemical evidence of CPHD, the diagnosis of acquired isolated idiopathic GH deficiency demands careful and thorough documentation. Indeed, recent data suggest that retesting of such patients may be associated with an early reversal of the diagnosis. The overdiagnosis of GH deficiency likely arises from the poor specificity of provocative GH testing, particularly in prepubertal children.
Bioinactive growth hormone
A small number of patients with a defect in the GH1 gene that generates a biologically inactive but immunoreactive GH molecule have been reported. These patients have a common phenotype and are characterized by severe short stature (usually height SDS <–3) and very low concentrations of IGF-1 and IGFBP-3, but normal or elevated GH concentrations (basal or after a stimulation test). Patients may present with mild clinical characteristics, such as prominent forehead and saddle nose, that are typical of GHI. Contrary to what would be expected in a patient with GHI, these patients showed some improvement in growth rate and IGF-1 levels with exogenous GH treatment. Missense mutations in GH1 , which are inherited in an autosomal dominant or recessive fashion, generate a mutated GH that is capable of binding to the receptor, but cannot properly activate signal transduction by the GHR.
Growth hormone insensitivity
GHI is a genetic disorder that is defined as the inability to respond to endogenous and exogenous GH with appropriate growth and metabolic effects. The classical form of GHI, also known as Laron syndrome , is characterized by severe short stature that manifests postnatally and is associated with dysmorphic features, such as craniofacial disproportion with a relatively small face, depressed nasal bridge, high-pitched voice, truncal obesity, and micropenis, a phenotype similar to that seen in patients with complete congenital GH deficiency ( Fig. 11.5 ). Typically, GHI patients show very low concentrations of IGF-1, IGFBP-3, and ALS, but elevated basal and stimulated GH concentrations. In most cases, GHI is caused by a fully penetrant autosomal recessive mutation in the GHR . The majority of these mutations are located in exons that encode the extracellular domain of GHR and disrupt the ligand-binding domain and/or impair transport of the receptor to the plasma membrane. For these reasons, patients with classic GHI often show low or undetectable GHBP levels because GHBP is generated by proteolytic cleavage of the extracellular GHR domain. However, mutations elsewhere in the GHR gene may result in normal or elevated GHBP concentrations.