The human myeloproliferative neoplasms (MPN) have long been associated with abnormal responses to cytokines and activation of signaling pathways, although the exact molecular mechanisms underlying these observations were unknown. This situation altered with the discovery of the JAK2 V617F, which presaged the ongoing description of further mutations predicted to activate canonical signaling pathways in MPN. This article covers the nature of these mutations and summarizes functional experiments in model systems and in human MPN cells to define the signaling pathways altered and how these drive and determine the MPN cellular phenotype. Also discussed are recently described, novel noncanonical signaling pathways to chromatin predicted to alter gene transcription more directly and to also contribute to the MPN phenotype.
- •
Human myeloproliferative neoplasms (MPN) are defined by abnormal signaling patterns, which are central to the development of these diseases.
- •
The studies conducted so far suggest that multiple signaling pathways are upregulated, with many linked to specific recurrent mutations.
- •
Although the data suggest the induction of common signaling pathways, emerging evidence also suggests that specific signaling pathways or combinations of signaling abnormalities may dictate the phenotypic differences among MPN subtypes.
- •
Further delineating these signaling pathways and how they might differ from homeostatic signaling pathways in normal hematopoietic stem and progenitor populations will greatly inform knowledge of MPN and allow clinicians to more safely target signaling for therapeutic gain.
Initial clues about altered signaling pathways in MPN
In 1974, Prchal and Axelrad demonstrated the seminal finding that bone marrow (BM) cells from patients with PV could form colony-forming units–erythroid in vitro without the addition of erythropoietin (EPO). These colonies were therefore called endogenous erythroid colonies (EECs). Because overproduction of red blood cells in PV is also associated with reduced EPO levels, it was proposed that EECs were generated by progenitors either hypersensitive to EPO or whose growth was EPO-independent. This observation was confirmed by culturing BM or peripheral blood (PB) progenitor cells in vitro in the presence of neutralizing antibodies to EPO or EPO-R and by replating analysis in methylcellulose media. Correa and coworkers went on to develop serum-free culture medium to study the growth of PV progenitors in vitro and found that erythroid progenitors were also hypersensitive to other cytokines including insulin-like growth factor-1, but not to EPO. Other groups demonstrated hypersensitivity of erythroid progenitors to several cytokines involved in myeloid development, including interleukin (IL)-3, granulocyte-macrophage colony–stimulating factor (GM-CSF), and stem cell factor in patients with PV compared with normal control subjects. By contrast, they did not observe any difference in response when PV progenitors were stimulated with cytokines with more pleiotrophic effects, including IL-4, IL-6, granulocyte colony–stimulating factor (G-CSF), and macrophage colony–stimulating factor (M-CSF). It was therefore concluded that erythroid or myeloid progenitor cells from patients with PV were hypersensitive to the predominant cytokines that instructed their proliferation and differentiation. Further experiments were performed to assess number, affinity, and internalization of I 125 -labeled EPO and IL-3 receptors in erythroid progenitors from patients with PV and normal control subjects and excluded differences as an explanation for altered cytokine sensitivity. Furthermore, no mutations were detected in the EPO receptor gene in patients with PV.
Mutations associated with activated signaling in patients with MPN
Although the previously mentioned studies demonstrated abnormal signaling in patients with MPN, the molecular basis for these findings was not elucidated for a further two decades. In contrast, over the last 7 years several mutations have been described that provide a framework for the understanding of activated signaling in MPN.
JAK2 V617F Mutation
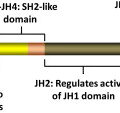
In 2005, several groups identified a somatic mutation (G-to-T substitution at position 1849) in the tyrosine kinase JAK2 in patients with MPN. This mutation occurs in most patients with PV (95%–99%), and in a significant proportion of patients with ET (50%–70%) and patients with IMF (40%–50%). The mutation results in the substitution of valine to phenylalanine at codon 617 (V617F). JAK2 is a member of the Janus family of cytoplasmic tyrosine kinases, which also includes JAK1, JAK3, and TYK2. JAK kinases contain two homologous kinase domains: a catalytically active JH1 domain and what was until recently thought to be an inactive JH2 domain ( Fig. 1 ). The V617F mutation alters the JH2 domain, which contains a similar amino acid sequence to JH1 and is also called the pseudokinase domain. The JH2 domain was already known to play a negative auto regulatory role in switching off the kinase activity of JAK2, because its deletion results in constitutive activation of the kinase. However, Ungureanu and colleagues have recently demonstrated that the JH2 pesudokinase domain also possesses catalytic activity and that it is this activity that regulates the JAK2 JH1 kinase activity. This occurs through auto-phosphorylation at two negative regulatory sites, one of which (Tyr570) had been previously shown to regulate response to cytokines.
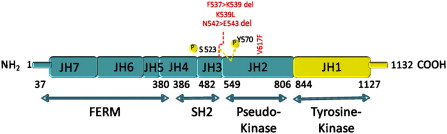
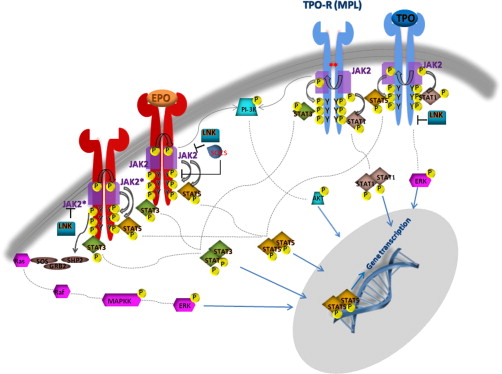
JAK2 is associated with the cytoplasmic tails of many cytokine receptors, including the type I cytokine receptors for EPO, TPO, G-CSF, GM-CSF, and IL-3, which are involved in myeloid or erythroid differentiation. In particular, JAK2 is the only kinase specifically associated with EPO-R and MPL (TPO-R) and is also involved, by its N terminal FERM domain, in facilitating the transport of the receptor-JAK complex to the cell surface. When a specific cytokine binds its cognate-receptor it induces receptor dimerization or oligomerization. JAK family members are preassembled on the receptor in the endoplasmic reticulum and ligand binding induces their transphosphorylation. The fully activated JAKs subsequently phosphorylate specific tyrosine residues on the receptor, which act as docking sites for STAT (signal transducers and activators of transcription) proteins and other SH2 domain-containing adaptor proteins ( Fig. 2 ). The STAT proteins are, in turn, themselves phosphorylated inducing their dimerization and translocation to the nucleus, where they activate the transcription of target genes, many of which are involved in cell proliferation. In addition to STAT proteins, JAK kinases can also activate other signaling pathways by binding SH2-containing adaptor proteins. These include RAS, mitogen-activated protein kinase-extracellular signal-related kinase (MAPK-ERK), protein kinase B (PKB or AKT), and phospholipase-B pathways. The MPN V617F mutation is thought to abrogate the catalytic activity of the JH2 domain, thereby increasing the basal activity of JAK2. In support of this notion, expression of JAK2 V617F in murine hematopoietic cells leads to cytokine hypersensitivity and cytokine-independent growth similar to EECs grown from patients with PV. Constitutive phosphorylation of JAK2 in the absence of ligand is also observed. Cytokine-independent growth seems most effective if the cells also express homodimeric type 1 cytokine receptors, such as EPO-R, MPL, or G-CSF-R. This is hardly surprising, in that these are receptors that are differentially expressed on cells of the erythroid, megakaryocytic, and granulocytic lineages, the lineages preferentially amplified in MPNs.
JAK2 Exon 12 Mutations
Approximately 5% of patients with PV and almost half of patients with ET and MF do not harbor the JAK2 V617F mutation. Therefore, further studies were undertaken to identify complementing alleles that might be mutated in these patients. Scott and colleagues analyzed JAK2V617F-negative patients with PV for mutations in all JAK kinase family members and their downstream mediators STAT5A and B. They found four novel mutations in exon 12 of JAK2 . Of the initial 10 patients analyzed, six had mutations that either involved deletion or substitution affecting lysine at codon 539 ( Table 1 ). The remaining four had N542-E543 deletion. Erythroid colonies could be grown from these patients in vitro in the absence of EPO, with all colonies heterozygous for the exon 12 mutation. However, unlike V617F, which can occur in the homozygous state, no colonies homozygous for exon 12 mutations were found. More recently, a large number of other JAK2 exon 12 mutations have been identified. Structurally, all of the mutations are present upstream of the JH2 pseudokinase domain (see Fig. 1 ), and are thought to modify the conformation of this domain, leading to an increased basal activity of JAK2. Unlike JAK2V617F, exon 12 mutations have not been found in patients with ET or PMF. Expression of mutant K539L JAK2 in murine Ba/F3 cells expressing the EPO-R led to factor-independent growth. However, no growth was observed in the absence of EPO-R, indicating that these mutants also required the presence of homodimeric type-1 cytokine receptors to manifest their effect. In addition, in a murine retroviral BM transplantation (BMT) assay model of MPN overexpressing the K539L JAK2 exon 12 mutant, the resultant myeloproliferative phenotype was distinct from V617F, suggesting that different mutations in the same kinase can lead to distinct clinical phenotypes, presumably through subtle differences in signaling.
| Function | Disease | Mutation | Frequency | References | |
|---|---|---|---|---|---|
| JAK2 | Tyrosine kinase | PV, ET, MF PV PV PV | V617F K539L F537-K539delinsL N542-E543del | PV (95%) ET, MF (∼50%) Exon 12 mutations: PV (5%) | |
| MPL | Thrombopoietin receptor | ET, MF | W515L W515K W515N W515A | ET (4%) MF (11%) | |
| LNK (SH2B3) | Adaptor protein; negative regulator of signaling | ET, MF | DEL mutant (loss of PH&SH2 domains) E208Q Other LNK mutations outside of PH&SH2 domains | ET (<5%) MF (<5%) | |
| CBL | Adaptor protein; E3 ubiquitin ligase; negative regulator of signaling | MF | Missense mutations in RING finger domain leading to loss of ubiquitin ligase activity Other CBL mutations in Proline rich domain no loss of ubiquitin ligase activity | MF (6%) | |
| SOCS3 | E3 ubiquitin ligase; negative regulator of signaling | PV | SH2 mutant F136L | PV (2.4% in a Japanese cohort) |
MPL Mutations
Several laboratories working on Tpo signaling had previously reported that overexpression of Tpo causes myelofibrosis in mice. In support of abnormal TPO-MPL signaling in MPN pathogenesis, gain-of-function somatic mutations in JAK2V617F-negative patients with PMF and ET have since been identified in MPL . No mutations have been documented in patients with PV. The first mutation reported was substitution of tryptophan to leucine at position 515 (W515L). The tryptophan is located in the cytoplasmic membrane proximal region of the receptor and is involved in repression of the receptor in the absence of ligand and the transforming activity of the mutant receptor is also dependent on its cell surface localization. Similarly to JAK2 mutants, expression of mutant MPL receptor leads to cytokine independent growth and hypersensitivity to Tpo in cell lines in vitro and mouse BM engineered to overexpress the mutant receptor generated myelofibrosis with marked thrombocytosis in recipient mice after transplantation. Subsequently, other mutations targeting the same amino acid have been identified (see Table 1 ).
LNK Mutations
LNK proteins have no intrinsic catalytic activity or transcriptional domain, but demonstrate the ability to integrate signaling events within the cell. Lnk belongs to the PSM family of adaptor proteins (proline-rich, pleckstrin homology [PH] domain and Src homology 2 [SH2] domain-containing partner of the insulin receptor) and has been demonstrated to negatively regulate signaling through the MPL and EPO receptors by reducing the duration of their response to cytokine stimulation. On cytokine stimulation, LNK binds by its SH2 domain to phosphorylated JAK2, leading to attenuation of signaling. LNK can also bind MPL by its SH2 domain and was found to be colocalized with MPL to the plasma membrane in transfected cell lines. As might be expected, LNK levels were found to be high in CD34 + cells from patients with MPN and LNK expression was induced in vitro on overexpression of mutant JAK2. However, mutant JAK2 and MPL seem equally sensitive to negative regulation by LNK compared with their wild-type (WT) counterparts. LNK mutations have recently been identified in patients with ET and PMF that were negative for JAK2V617F, JAK2 exon 12, and MPLW515 mutations. Both mutations were located in exon 2 of LNK. One of the mutations led to loss of PH and SH2 domains, whereas the other was a point mutation in the PH domain (see Table 1 ). More recently, around 14 LNK mutations have been detected in patients with MPN. These mutations are not restricted to the PH or SH2 domains, have also been found to co-occur in JAK2V617F-positive patients, and may facilitate progression of the MPN, as suggested by their lower incidence in chronic-phase MPN (<10% mutated) and increasing incidence in accelerated and blast phase MPN. In vitro experiments in Ba/F3 MPL-expressing cells demonstrated that the SH2 domain LNK mutant could not inhibit TPO-dependent growth of these cells in contrast to WT LNK. The PH domain mutant was intermediate and partially retained its inhibitory effect. In addition, in BMT assays Lnk deficiency led to an accelerated onset of myeloproliferative disease induced by JAK2V617F, indicating that WT LNK inhibits oncogenic JAK2 activity. LNK −/− mice exhibit a subtle MPN phenotype in the absence of mutant JAK2 expression, with an enlarged stem cell compartment suggestive of the fact that LNK plays a role in pathogenesis of MPN.
CBL Mutations
CBL is the cellular homolog of the v-Cbl oncogene, which is the transforming gene of Casitas B lymphoma murine retrovirus. CBL proteins have a multidomain structure with a unique amino terminal tyrosine kinase binding domain, a linker region, and a RING finger domain. The C terminus of CBL contains tyrosine residues, which on phosphorylation interact with SH2 domain proteins. It also contains Proline rich regions, which interact with SH3 domain proteins. The main role of CBL proteins is as negative regulators of signaling by different tyrosine kinases (receptor and nonreceptor). The mechanism of action is by their RING finger domain, which is an E3 ubiquitin ligase that targets activated tyrosine kinases for degradation by ubiquitination. CBL can also target cytokine receptors, including MPL, for degradation. In classical MPNs, CBL mutations were first detected in JAK2 mutation–negative patients with PMF with an acquired uniparental disomy of the 11q region, in which the CBL gene lies. The mutations were missense mutations involving the RING finger and linker regions that abrogated ubiquitin ligase activity and led to autonomous growth of 32D/FLT3 cells in vitro. More recently, mutations have also been detected in JAK2V617F-positive MPNs. These involved the proline-rich region of CBL and would be predicted not to affect the ubiquitination activity of CBL. However, in vitro these mutations do lead to factor-independent growth, although the mechanisms remain unclear. Interestingly, CBL mutations are more frequent in PMF compared with ET and PV, and similarly to LNK mutations, acquisition of mutations in CBL may be late events in MPN, potentially heralding their transformation to acute myeloid leukemia.
Suppressors of Cytokine Signaling Alterations
Suppressors of cytokine signaling (SOCS) proteins were initially described as inhibitory regulators of the JAK-STAT signaling pathway. The SOCS family consists of eight intracellular proteins that exert their negative feedback through several mechanisms. Among the SOCS family members, SOCS1 and SOCS3 compete with STAT proteins to bind to the JAK2 catalytic domain resulting in abrogation of downstream signaling. They can also bind by their SH2 domains to phospho-tyrosines on activated cytokine receptors (eg, EPO-R), thus preventing JAK2 association with the receptor. SOCS proteins are also components of the ubiquitin E3 ligase family and target their binding partners, including activated JAKs, for ubiquitination and degradation. SOCS3 germline mutation (F316L) has been detected in a small cohort of Japanese patients and in a patient with a heterozygous JAK2 exon 12 mutation. The mutation impairs the degradation of the SOCS-JAK complex after ubiquitination. However, because the SOCS3 mutation is a relatively rare event, its role in disease pathogenesis is unclear. In addition, other mechanisms that impair SOCS expression have been described, including hypermethylation of CpG islands near the promoters of SOCS1 and SOCS-3 in patients with MPN. Furthermore, functional studies into the role of SOCS proteins in the regulation of WT and mutant JAK2 are somewhat conflicting. In particular, one study suggests an inability of SOCS3 to negatively regulate mutant JAK2 and a stabilization of SOCS3 by JAK2-mediated phosphorylation. However, these findings were not corroborated in another study, highlighting the complex role of SOCS proteins in modulating abnormal signaling within patients with MPN.
Mutations associated with activated signaling in patients with MPN
Although the previously mentioned studies demonstrated abnormal signaling in patients with MPN, the molecular basis for these findings was not elucidated for a further two decades. In contrast, over the last 7 years several mutations have been described that provide a framework for the understanding of activated signaling in MPN.
JAK2 V617F Mutation
In 2005, several groups identified a somatic mutation (G-to-T substitution at position 1849) in the tyrosine kinase JAK2 in patients with MPN. This mutation occurs in most patients with PV (95%–99%), and in a significant proportion of patients with ET (50%–70%) and patients with IMF (40%–50%). The mutation results in the substitution of valine to phenylalanine at codon 617 (V617F). JAK2 is a member of the Janus family of cytoplasmic tyrosine kinases, which also includes JAK1, JAK3, and TYK2. JAK kinases contain two homologous kinase domains: a catalytically active JH1 domain and what was until recently thought to be an inactive JH2 domain ( Fig. 1 ). The V617F mutation alters the JH2 domain, which contains a similar amino acid sequence to JH1 and is also called the pseudokinase domain. The JH2 domain was already known to play a negative auto regulatory role in switching off the kinase activity of JAK2, because its deletion results in constitutive activation of the kinase. However, Ungureanu and colleagues have recently demonstrated that the JH2 pesudokinase domain also possesses catalytic activity and that it is this activity that regulates the JAK2 JH1 kinase activity. This occurs through auto-phosphorylation at two negative regulatory sites, one of which (Tyr570) had been previously shown to regulate response to cytokines.
JAK2 is associated with the cytoplasmic tails of many cytokine receptors, including the type I cytokine receptors for EPO, TPO, G-CSF, GM-CSF, and IL-3, which are involved in myeloid or erythroid differentiation. In particular, JAK2 is the only kinase specifically associated with EPO-R and MPL (TPO-R) and is also involved, by its N terminal FERM domain, in facilitating the transport of the receptor-JAK complex to the cell surface. When a specific cytokine binds its cognate-receptor it induces receptor dimerization or oligomerization. JAK family members are preassembled on the receptor in the endoplasmic reticulum and ligand binding induces their transphosphorylation. The fully activated JAKs subsequently phosphorylate specific tyrosine residues on the receptor, which act as docking sites for STAT (signal transducers and activators of transcription) proteins and other SH2 domain-containing adaptor proteins ( Fig. 2 ). The STAT proteins are, in turn, themselves phosphorylated inducing their dimerization and translocation to the nucleus, where they activate the transcription of target genes, many of which are involved in cell proliferation. In addition to STAT proteins, JAK kinases can also activate other signaling pathways by binding SH2-containing adaptor proteins. These include RAS, mitogen-activated protein kinase-extracellular signal-related kinase (MAPK-ERK), protein kinase B (PKB or AKT), and phospholipase-B pathways. The MPN V617F mutation is thought to abrogate the catalytic activity of the JH2 domain, thereby increasing the basal activity of JAK2. In support of this notion, expression of JAK2 V617F in murine hematopoietic cells leads to cytokine hypersensitivity and cytokine-independent growth similar to EECs grown from patients with PV. Constitutive phosphorylation of JAK2 in the absence of ligand is also observed. Cytokine-independent growth seems most effective if the cells also express homodimeric type 1 cytokine receptors, such as EPO-R, MPL, or G-CSF-R. This is hardly surprising, in that these are receptors that are differentially expressed on cells of the erythroid, megakaryocytic, and granulocytic lineages, the lineages preferentially amplified in MPNs.
JAK2 Exon 12 Mutations
Approximately 5% of patients with PV and almost half of patients with ET and MF do not harbor the JAK2 V617F mutation. Therefore, further studies were undertaken to identify complementing alleles that might be mutated in these patients. Scott and colleagues analyzed JAK2V617F-negative patients with PV for mutations in all JAK kinase family members and their downstream mediators STAT5A and B. They found four novel mutations in exon 12 of JAK2 . Of the initial 10 patients analyzed, six had mutations that either involved deletion or substitution affecting lysine at codon 539 ( Table 1 ). The remaining four had N542-E543 deletion. Erythroid colonies could be grown from these patients in vitro in the absence of EPO, with all colonies heterozygous for the exon 12 mutation. However, unlike V617F, which can occur in the homozygous state, no colonies homozygous for exon 12 mutations were found. More recently, a large number of other JAK2 exon 12 mutations have been identified. Structurally, all of the mutations are present upstream of the JH2 pseudokinase domain (see Fig. 1 ), and are thought to modify the conformation of this domain, leading to an increased basal activity of JAK2. Unlike JAK2V617F, exon 12 mutations have not been found in patients with ET or PMF. Expression of mutant K539L JAK2 in murine Ba/F3 cells expressing the EPO-R led to factor-independent growth. However, no growth was observed in the absence of EPO-R, indicating that these mutants also required the presence of homodimeric type-1 cytokine receptors to manifest their effect. In addition, in a murine retroviral BM transplantation (BMT) assay model of MPN overexpressing the K539L JAK2 exon 12 mutant, the resultant myeloproliferative phenotype was distinct from V617F, suggesting that different mutations in the same kinase can lead to distinct clinical phenotypes, presumably through subtle differences in signaling.
| Function | Disease | Mutation | Frequency | References | |
|---|---|---|---|---|---|
| JAK2 | Tyrosine kinase | PV, ET, MF PV PV PV | V617F K539L F537-K539delinsL N542-E543del | PV (95%) ET, MF (∼50%) Exon 12 mutations: PV (5%) | |
| MPL | Thrombopoietin receptor | ET, MF | W515L W515K W515N W515A | ET (4%) MF (11%) | |
| LNK (SH2B3) | Adaptor protein; negative regulator of signaling | ET, MF | DEL mutant (loss of PH&SH2 domains) E208Q Other LNK mutations outside of PH&SH2 domains | ET (<5%) MF (<5%) | |
| CBL | Adaptor protein; E3 ubiquitin ligase; negative regulator of signaling | MF | Missense mutations in RING finger domain leading to loss of ubiquitin ligase activity Other CBL mutations in Proline rich domain no loss of ubiquitin ligase activity | MF (6%) | |
| SOCS3 | E3 ubiquitin ligase; negative regulator of signaling | PV | SH2 mutant F136L | PV (2.4% in a Japanese cohort) |
Related posts:
 Genotype-Phenotype Interactions in the Myeloproliferative Neoplasms
Genotype-Phenotype Interactions in the Myeloproliferative Neoplasms
 Role of TET2and ASXL1Mutations in the Pathogenesis of Myeloproliferative Neoplasms
Role of TET2and ASXL1Mutations in the Pathogenesis of Myeloproliferative Neoplasms
 Systemic Mastocytosis
Systemic Mastocytosis
 Acquired Uniparental Disomy in Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
Acquired Uniparental Disomy in Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
 Therapy with JAK2 Inhibitors for Myeloproliferative Neoplasms
Therapy with JAK2 Inhibitors for Myeloproliferative Neoplasms
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


