Since the discovery of activating mutations in JAK2 in patients with myeloproliferative neoplasms (MPNs) in 2005, gene discovery efforts have identified additional disease alleles, which can predate or occur subsequent to acquisition of JAK2/MPL mutations. In 2009, somatic copy number loss and mutations in the genes TET2 and ASXL1 were identified in MPN patients. Genetic analysis of MPN patient cohorts with adequate sample size and clear clinical annotation are needed to understand the importance of these mutations on MPN phenotype, risk of transformation to leukemia, response to therapy, and influence on overall survival.
- •
Mutations in TET2 are loss-of-function mutations present in 9% to 16% of patients with polycythemia vera (PV), 4% to 5% of patients with essential thrombocytosis (ET), and 7% to 17% of patients with primary myelofibrosis (PMF) or myelofibrosis arising from PV or ET.
- •
TET2 is a member of the TET family of α-ketoglutarate and Fe(II)-dependent dioxygenases, which oxidize 5-methylcytosine on DNA to produce 5-hydroxymethylcytosine followed by 5-formylcytosine and 5-carboxylcytosine. This enzymatic activity of TET2 is thought to facilitate demethylation of DNA.
- •
Additional sex combs like 1 (ASXL1) is the mammalian homologue of Drosophila ASXL, a protein known to affect both Trithorax group and Polycomb group gene function. Although several functions have been ascribed to ASXL1 in nonmammalian and nonhematopoietic cell contexts, the function of ASXL1 in mammalian hematopoietic cells is not yet fully delineated.
- •
Mutations in ASXL1 are most common amongst MPN patients with PMF (13%–26%) or post-PV/ET MF (22%–38%) as compared with patients with PV (2%–5%) or ET (5%–10%).
- •
The clinical importance of TET2 and ASXL1 mutations amongst patients with MPNs is not yet clear. ASXL1 mutations may confer worsened overall survival amongst patients with PMF/post-PV/ET MF, but this needs to be validated in larger, prospective studies.
Evidence for mutations outside of the JAK-STAT pathway in patients with myeloproliferative neoplasms
Myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoiesis characterized by excess production of mature-appearing cells within the blood stream. The MPNs were initially grouped together by William Dameshek in 1951. However, in 2005, the first biologic basis unifying the pathogenesis of the different MPNs was discovered when activating mutations in JAK2 were identified in 95% of patients with polycythemia vera (PV), 55% to 60% of patients with essential thrombocytosis (ET), and 50% of patients with primary myelofibrosis (PMF). This discovery was quickly followed by the discovery of additional mutations, resulting in activation of the JAK-STAT pathway in MPN patients, including exon 12 mutations in JAK2 , thrombopoietin receptor ( MPL) mutations, and loss-of-function mutations in LNK , a negative regulator of JAK-STAT signaling ( Table 1 ).
| Gene Mutation a | ||||||
|---|---|---|---|---|---|---|
| MPN | JAK2 V617F | JAK2 Exon 12 | MPL | LNK | TET2 | ASXL1 b |
| Polycythemia vera (PV) | 95% | 3%–5% | NR | Present | 9.8%–16% | 2%–5% |
| Essential thrombocytosis (ET) | 55%–60% | NR | 3%–5% | 3%–6% | 4.4%–5% | 5%–10% |
| Primary myelofibrosis (MF) | 50%–60% | NR | 8%–10% | 3%–6% | 7.7%–17% | 13%–26% |
| Post-PV/ET MF | 50%–60% | NR | NR | NR | 14% | 22%–38.5% |
b Several manuscripts used to delineate mutational frequency of ASXL1 contain the controversial p.Gly646TrpfsX12 variant which has not definitively proven to be a somatic mutation.
Although the discovery of mutations in JAK2 and MPL provided seminal insight into MPN pathogenesis, several lines of evidence suggest that mutations in genes other than JAK2 and MPL must be present in MPN patients. First was the question of how a single mutation in JAK2, which appeared to be sufficient for MPN pathogenesis from in vivo studies, could result in the development of 3 phenotypically different diseases. One attractive hypothesis to this question was that additional acquired or inherited genetic modifiers outside of JAK2 or MPL could be present and modify the MPN phenotype. Secondly, clonal analysis of patients with JAK2/MPL mutations demonstrated the presence of JAK2 wild-type endogenous erythroid colonies, clear evidence that an additional aberration responsible for erythropoietin-independent growth must be present. Clonality analysis of patients with a cytogenetic abnormality in conjunction with the JAK2V617F mutation also revealed that some patients had cytogenetically abnormal clones with and without the JAK2V617F mutation. Finally, several reports have shown that leukemic blasts of acute myeloid leukemia (AML) derived from a JAK2V617F MPN are frequently JAK2 wild-type, suggesting that the MPN and AML clones can arise from 2 different progenitor cells or that an ancestral clone bearing an abnormality preceding the JAK2V617F mutation can give rise to both the JAK2-positive MPN and the JAK2-negative AML. Thus, additional novel mutations in MPN pathogenesis have been speculated to exist since the discovery of mutations in JAK2 .
Discovery of TET2 mutations in MPN patients
Mutations in TET2 (Ten-Eleven-Translocation 2) were the first described recurrent somatic alterations in MPN patients in a gene not directly known to be involved in the JAK-STAT signaling pathway. TET2 mutations were originally described by Delhommeau and colleagues and Langemeijer and colleagues in 2009 in patients with MPNs and myelodysplastic syndrome. Through careful examination of primary PV patient samples, Delhommeau and colleagues noticed that most of JAK2V617F mutant PV patients (∼85%) had expansion of CD34+CD38+––committed progenitor cells over CD34+CD38− multipotent progenitors in ex vivo liquid cultures. In contrast, a minority (∼15%) of JAK2V617F mutant PV patients were characterized by relative expansion of the more immature multipotent progenitor cells (CD34+ CD38−). Hypothesizing that a novel genetic abnormality might be responsible for this immunophenotypic difference in these 2 patient subsets, the investigators performed single nucleotide polymorphism arrays (Affymetrix 500K) and comparative genomic hybridization array (aCGH) (Agilent 244K) on a small number of patient samples. They found that 3 of the 5 JAK2V617F mutant PV patients with expansion of CD34+CD38− cells had loss of heterozygosity at chromosomal locus 4q24. One of these 5 patients had a deletion of a 325 kB region of DNA at 4q24, the only gene present in this region being TET2. This deletion then led to sequencing of TET2 in these patient samples and the identification of somatic TET2 mutations in MPNs. Since then, sequencing of TET2 has led to the identification of TET2 mutations in every myeloid disorder. Mutations in TET2 have been found in all coding regions and can appear as missense, nonsense, or frameshift mutations. Mutations in TET2 are less uncommonly biallelic (ie, involving both copies of TET2 ), consistent with mutations in TET2 being haploinsufficient loss-of-function mutations in most patients.
Discovery of TET2 mutations in MPN patients
Mutations in TET2 (Ten-Eleven-Translocation 2) were the first described recurrent somatic alterations in MPN patients in a gene not directly known to be involved in the JAK-STAT signaling pathway. TET2 mutations were originally described by Delhommeau and colleagues and Langemeijer and colleagues in 2009 in patients with MPNs and myelodysplastic syndrome. Through careful examination of primary PV patient samples, Delhommeau and colleagues noticed that most of JAK2V617F mutant PV patients (∼85%) had expansion of CD34+CD38+––committed progenitor cells over CD34+CD38− multipotent progenitors in ex vivo liquid cultures. In contrast, a minority (∼15%) of JAK2V617F mutant PV patients were characterized by relative expansion of the more immature multipotent progenitor cells (CD34+ CD38−). Hypothesizing that a novel genetic abnormality might be responsible for this immunophenotypic difference in these 2 patient subsets, the investigators performed single nucleotide polymorphism arrays (Affymetrix 500K) and comparative genomic hybridization array (aCGH) (Agilent 244K) on a small number of patient samples. They found that 3 of the 5 JAK2V617F mutant PV patients with expansion of CD34+CD38− cells had loss of heterozygosity at chromosomal locus 4q24. One of these 5 patients had a deletion of a 325 kB region of DNA at 4q24, the only gene present in this region being TET2. This deletion then led to sequencing of TET2 in these patient samples and the identification of somatic TET2 mutations in MPNs. Since then, sequencing of TET2 has led to the identification of TET2 mutations in every myeloid disorder. Mutations in TET2 have been found in all coding regions and can appear as missense, nonsense, or frameshift mutations. Mutations in TET2 are less uncommonly biallelic (ie, involving both copies of TET2 ), consistent with mutations in TET2 being haploinsufficient loss-of-function mutations in most patients.
Biochemical function of TET2
TET2 is a member of the TET family of genes, the first member of which to be described was TET1 (Ten-Eleven-Translocation 1). TET1 , located on chromosome 10, was originally identified in cases of adult and pediatric AML as a translocation partner with MLL (located on chromosome 11). Although TET1 was the original gene member identified in hematologic malignancies, no sequence alterations in TET1 or TET3 have been identified to date.
The function of TET1 was first described in a landmark publication by Tahiliani and colleagues in 2009. They pursued the identification of human enzymes that modify bases of nucleic acids as a means to understand how catalytic modifications of DNA bases affect the genetic code. As such, they undertook in silico approach to identify human homologues of the trypanosome proteins JBP1 and JBP2, which are known to oxidize the 5-methyl group of thymine. Such enzymes were not previously known to exist in higher organisms. Surprisingly, they found that the TET family of genes was human homologues of these trypanosomal enzymes. Further characterization of TET1 revealed that it is a 2-oxoglutarate- and Fe(II)––dependent dioxygenase that serves to oxidize the 5-methyl group of cytosine, leading to formation of 5-hydroxymethylcytosine ( Fig. 1 ).
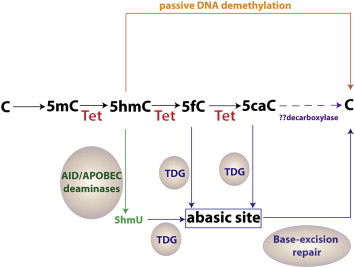
More recent work has identified that all 3 TET proteins are enzymes that can convert 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC). Moreover, 2 groups have reported that TET proteins can further convert 5hmC into 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) in 2 successive oxidation reactions (see Fig. 1 ).
The discovery of these novel enzymatic activities by the TET proteins has provided insight into potential mechanisms by which 5mC is dynamically regulated as well as demethylated in both active and passive processes. For instance, DNMT1 (DNA methyltransferase 1), the DNA methyltransferase responsible for maintaining DNA methylation, does not recognize 5hmC. Thus, conversion of 5mC into 5hmC may lead to replication-dependent passive demethylation of DNA (see Fig. 1 ). Furthermore, oxidized derivatives of 5hmC may serve in a replication-independent, active DNA demethylation process. Proof for this concept comes from the finding that thymine DNA glycosylase (TDG) can excise 5fC or 5caC in the context of CpG sites (TDG has minimal activity toward 5hmC). The resulting abasic site following TDG-excision can then be repaired by the base-excision repair (BER) pathway to generate unmethylated cytosines. There is also evidence that 5hmC can be actively deaminated into 5-hydroxymethyluracil (5hmU) by the activation-induced deaminase/apolipoprotein B mRNA editing enzyme complex. The resulting 5hmU could then be removed via the action of DNA glycosylases and the BER pathway. Finally, Ito and colleagues proposed that 5mC might be converted to cytosine simply by iterative oxidation of 5hmC by TET enzymes followed by a single decarboxylation of 5caC to regenerate cytosine by a yet unidentified putative decarboxylase. Although no decarboxylase capable of removing the carboxyl group from 5caC has been identified, this latter mechanism of iterative 5mC oxidation followed by decarboxylation is attractive in its simplicity, and no DNA repair mechanism is required to effect DNA demethylation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


