Diffuse Parenchymal Lung Disease: Introduction
The lung brings the ambient air that we breathe into close proximity with the systemic circulation. This allows for its essential function in gas exchange. However, this also exposes the lung to a variety of potentially injurious infectious and noninfectious environmental agents. The normal host response to such insults is to eradicate the etiologic agent and to repair the injury caused either directly by the agent or indirectly by the associated inflammatory process. This complex but well orchestrated and tightly regulated host response leads to eventual resolution of injury and restoration of normal lung architecture and function in most cases. However, if the inflammatory and/or repair response is dysregulated, a chronic alveolar/interstitial remodeling process with varying degrees of inflammation and fibrosis ensues. Damage to the pulmonary vascular endothelium via the circulation (e.g., drugs, systemic rheumatic disorders) may produce similar inflammatory/fibrotic reactions in the lung. This results in the restrictive physiology and gas-exchange abnormalities characteristic of the diffuse parenchymal lung diseases (DPLDs). When all potential etiologic agents and associations are considered, the list of DPLDs includes over 150 different clinical entities (Table 84-1). Thus, DPLDs comprise a large and heterogeneous group of diseases that are grouped into a single category based on common features in their clinical, radiographic, and physiological presentations.
|
|
|
|
|
|
|
|
The most prevalent and devastating of all the DPLDs is idiopathic pulmonary fibrosis (IPF), a chronic fibrotic disease of unknown etiology. IPF is primarily a disease of elderly patients. Elderly patients maybe particularly susceptible to this disease because of inherent deficiencies in immune function and/or in their inability to mount an appropriate repair response. Alternatively, IPF may present in elderly patients as a result of accumulated insults or because early manifestations of the disease are difficult to recognize. In this chapter, we will focus on emerging new concepts regarding IPF pathogenesis and the challenges facing clinicians taking care of these patients. We will also discuss other selected DPLDs that are relatively more common in the geriatric population.
Epidemiology
The prevalence of DPLD is estimated at about 20 to 40 per 100 000 in the United States DPLD accounts for 100 000 hospital admissions yearly. The increased use of pneumotoxic drugs to treat malignant and cardiovascular diseases in the elderly as well the rising median age of the U.S. population contribute to an increased incidence.
IPF has been reclassified into a group of diseases designated as the “idiopathic interstitial pneumonias” (IIPs) (Table 84-1). IIPs are rare in children, but increase with advancing age. IPF, the most common of the IIPs, has a prevalence of 6 to 14.6 per 100 000 persons in different series. In patients older than the age of 75 years, however, the prevalence may exceed 175 per 100 000. The median age of onset of IPF ranges from 50 to 70 years and the diagnosis is almost never made in a patient aged 40 years or younger. In contrast, other IIPs such as nonspecific interstitial pneumonia (NSIP) appear to occur in younger patients.
Prognosis and Natural History
DPLDs represent a group of clinical syndromes with varying histopathological patterns ranging from predominantly inflammatory to more fibrotic tissue reactions. This is well illustrated by the IIPs that comprise primarily inflammatory tissue reactions such as desquamative interstitial pneumonia (DIP) and NSIP, in contrast to fibrotic histopathological patterns such as usual interstitial pneumonia (UIP). The histopathological subtype has important clinical and prognostic significance. UIP, the histopathological correlate of IPF, carries the worst prognosis and is, in general, unresponsive to anti-inflammatory and immune-modulating therapies. The finding of UIP on lung biopsy predicts a median survival of less than 5 years. Other baseline characteristics that predict a poor prognosis include a low DLCO, increased A–a gradient, desaturation during a 6-minute walk test (SaO2 < 88%), pulmonary arterial hypertension (mPAP >25 mm Hg), and the finding of honeycombing on high-resolution computed tomography (HRCT). Dynamic predictors of poor prognosis include a decline in forced vital capacity (FVC) ≥ 10 (% predicted) over a 6-month period, decline in DLCO ≥ 15 (% predicted) over a 12-month period and serial changes in dyspnea score.
The precise relationships between different histopathological subtypes are not well defined. For example, it is not known if NSIP precedes UIP in the natural history of the disease or if they represent distinct tissue reaction patterns to an unknown injury in patients with varying host genetic/susceptibility factors (see section on “Pathogenesis”). With regard to the natural history of IPF, there is significant heterogeneity in the patient population. While the majority of patients experience a steady and inexorable decline in lung function, a significant proportion of patients with IPF succumb to acute exacerbations of the disease. The etiology and pathogenesis of these acute exacerbations are currently unknown.
Pathogenesis
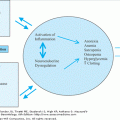
The pathogenesis of the DPLDs is complex. Significant differences in pathogenesis likely exist among the histopathological subtypes of IIP. Since UIP is also the most common form of IIP and since UIP/IPF exhibits an age-dependent increase in prevalence, we will focus on the pathogenesis of this clinical syndrome. IPF is localized to the lung parenchyma. Early histopathological evidence of alveolar epithelial cell injury and apoptosis suggests an inhaled route of entry for a putative injurious agent and/or an intrinsic abnormality specific to the alveolar epithelial cell. The temporal and spatial heterogeneity of the fibrotic lesions in UIP suggests that the injury maybe recurrent and repetitive, occurring over many years. Although the identity of a specific extrinsic agent has not been identified, latent viral infections and environmental toxins, including cigarette smoke, have been implicated. Alternatively, or in concert, intrinsic defects in alveolar epithelial cell function and fate may drive the fibrogenic tissue response (Figure 84-1).
Figure 84-1.
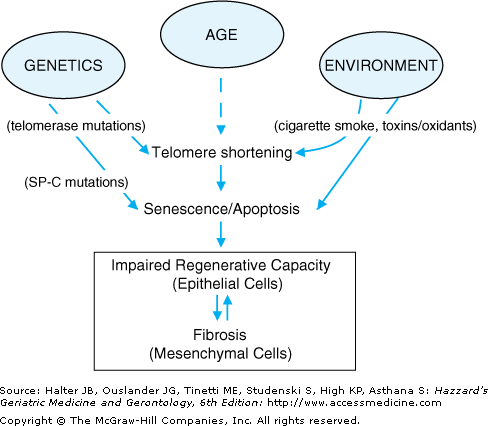
Host and environmental factors influence epithelial cell regenerative capacity. Mutations in the telomerase and surfactant protein-C (SP-C) genes have been linked to familial idiopathic pulmonary fibrosis; these telomerase and SP-C mutations induce cellular senescence and injury, respectively, leading to increased susceptibility of epithelial cells to apoptosis. Advancing age and extrinsic oxidative stress, including cigarette smoke, is also associated with telomere shortening and cellular senescence. Apoptosis of epithelial cells results in impaired re-epithelialization, loss of alveolar barrier function and activation of the underlying mesenchyme that culminates in fibrosis. The activated mesenchyme, in turn, may contribute to alveolar epithelial cell “dropout” by release of soluble mediators, further promoting epithelial dysrepair.
There is an increasing recognition that the aging process per se and the biology of cellular senescence influence tissue repair responses. Cellular senescence may affect the function and behavior of alveolar epithelial cells. Senescent epithelial cells with shortened telomeres are generally more susceptible to apoptosis. Genetic mutations in telomerase and surfactant protein-C have been shown to be associated with familial cases of IPF, suggesting a link to intrinsic defects in alveolar epithelial cell function. Familial pulmonary fibrosis appears to be inherited as an autosomal dominant trait with variable penetrance. No systematic assessment has been made of the heritability of IPF in the general population or the risk to relatives of affected individuals. Various forms of fibrotic lung disease have been associated with gene polymorphisms of interleukin-1-receptor antagonist, tumor necrosis-α, and major histocompatability complex loci. Transforming growth factor-β1 gene polymorphisms resulting in increased production of this cytokine have been associated with more rapid progression of the disease in patients with established IPF. Thus, aging and host genetic factors are likely to determine the susceptibility to fibrotic sequelae and, perhaps, in the type of pathologic repair that ensues in sporadic cases of fibrotic lung disease.