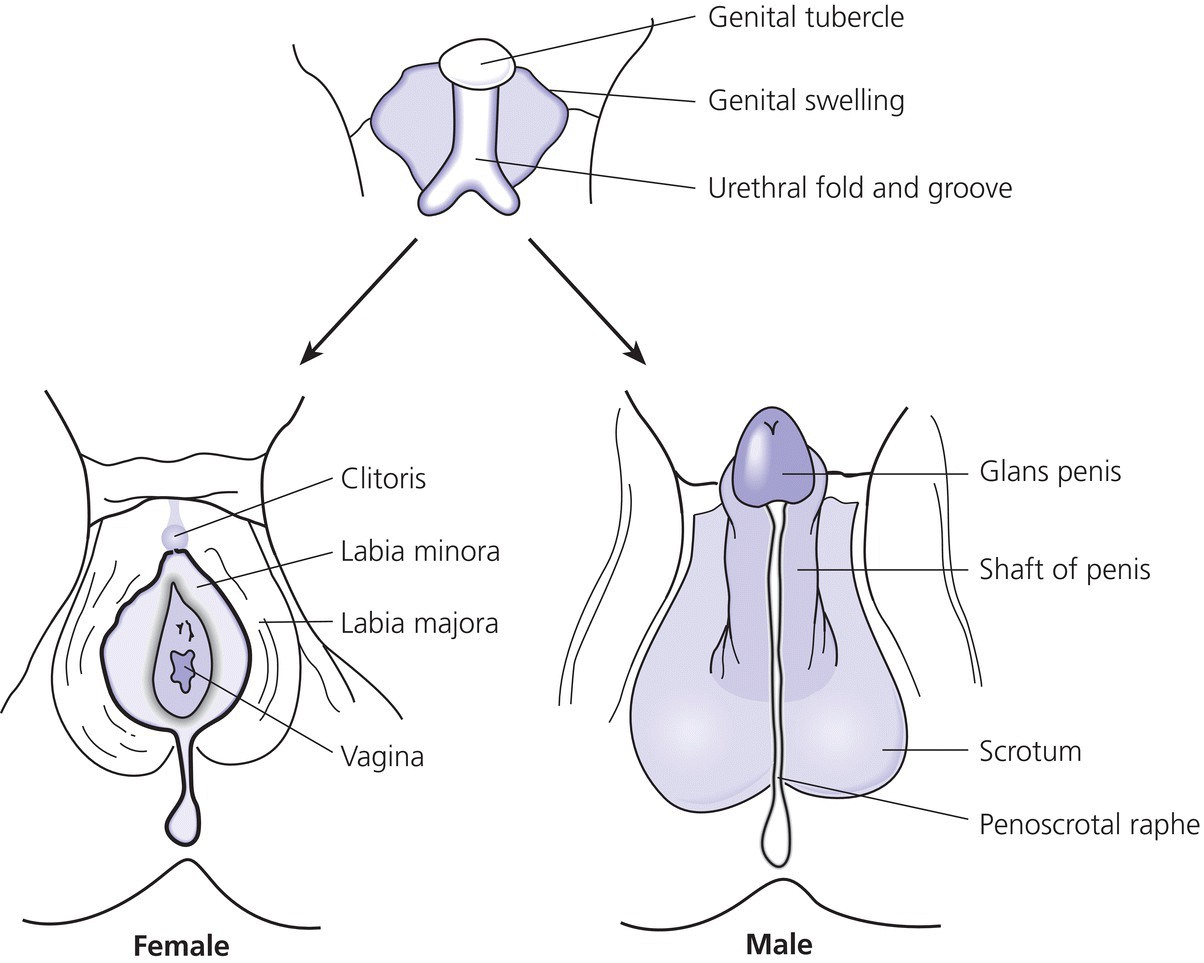
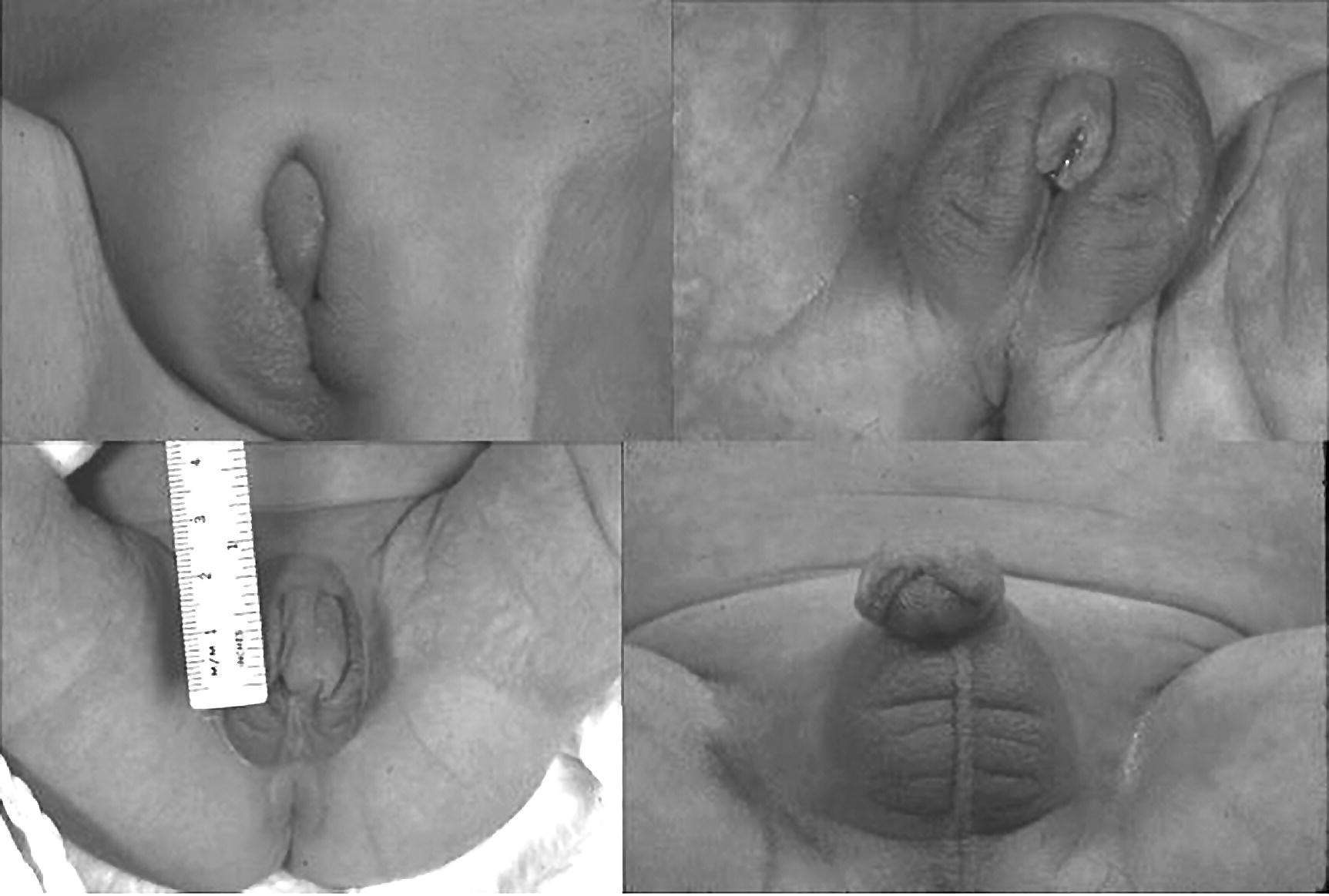
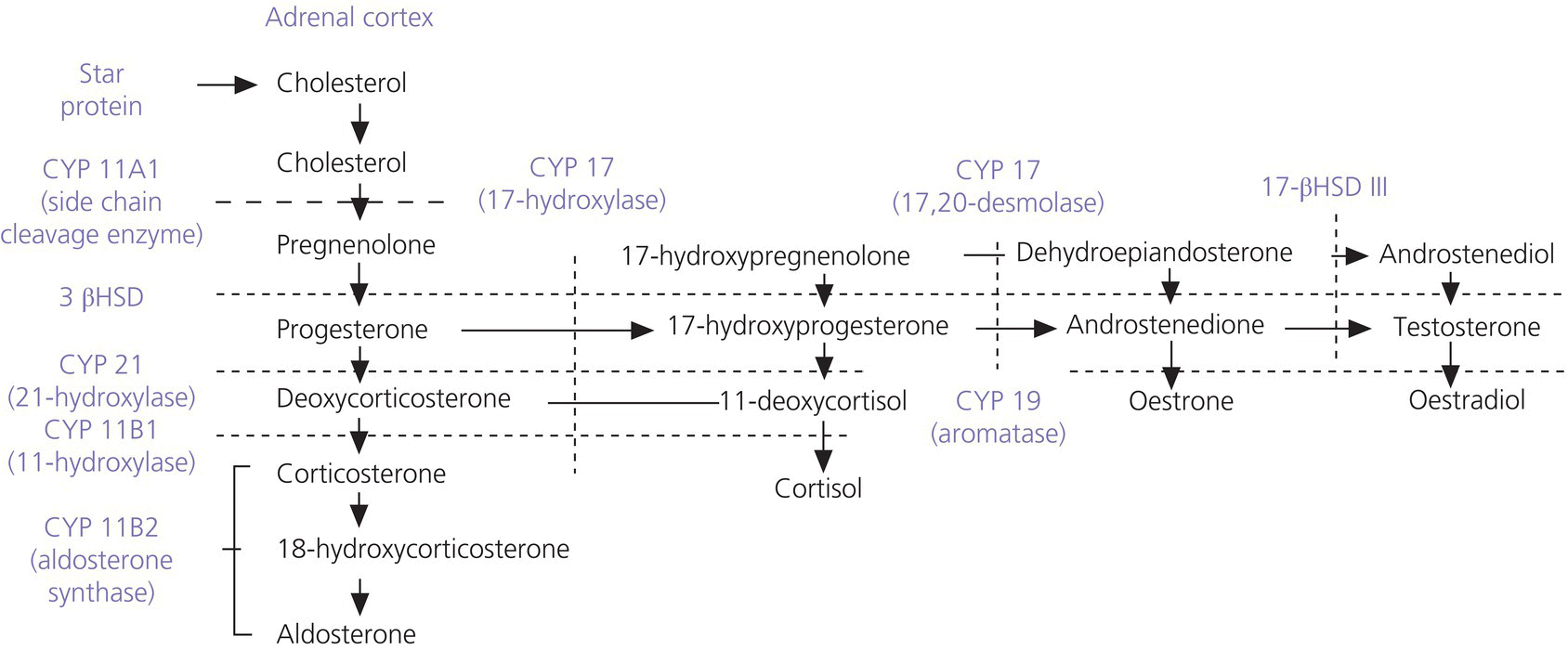
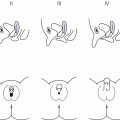
7 In 2006, the term ‘disorders of sex development (DSDs)’ was proposed to replace ‘intersex’, which was felt to be derogatory. Along the same lines, but to keep the same acronym, the term ‘differences’ can now be used instead of ‘disorder’, thus, differences in sex development. In the restricted sense, people with DSDs present with external genitalia that are sufficiently ambiguous to cause uncertainty as to which sex should be assigned and this is the major focus of this chapter. However, in the broader sense, DSDs encompass all situations when the karyotype is at partial or complete variance with the primary or secondary sex characteristics. Understanding normal sex differentiation and its genetic and hormonal control is a prerequisite to properly understand and manage people with DSDs. Perhaps even more so than in other fields of paediatrics, a basic understanding of the usefulness and limitations of the various cytogenetic and molecular tools available to the practitioner is important (see Chapter 13). Practitioners also need to be cognizant of the rapidly evolving social norms and values regarding sex and of the cultural differences in the way families react to and deal with a child with a DSD. The vast majority of people with gender incongruence have typical male or female genitalia and are not discussed in this chapter (see Further Reading for recent guidelines). However, having a DSD may increase the probability of a change in gender identity. An essential concept is that the human embryo is bipotential until 40 days gestation. Regardless of whether they are derived from a 46,XX or a 46,XY zygote, the gonads and the internal (Müllerian and Wolffian ducts) and external genitalia (genital tubercle and urethral and genital folds) have a similar appearance. After 40 days, the gonads differentiate into either testes, which start producing anti‐Müllerian hormone (AMH), a glycoprotein, at about six to eight weeks, and then testosterone, or into ovaries. Recent studies have shown that the differentiation of both the ovaries and the testes requires activation of specific gene cascades and that the ovaries actively inhibit Wolffian duct stabilization, thus challenging the long‐held concept of ‘female development by default’. However, this concept still applies to the differentiation of the external genitalia and of the urogenital sinus (see section ‘Urogenital Sinus’). A detailed discussion of this complex process is beyond the scope of this chapter. With few exceptions, the presence of a Y chromosome containing a normal SRY (sex‐determining region of the Y chromosome) gene will result in the differentiation of the bipotential gonad into a testis. However, a number of other genes, acting upstream or downstream of SRY, are required for normal testicular development. As noted above, some genes are also actively involved in the differentiation of the ovary. The list of these genes constantly expanding (see Further Reading). From six to eight weeks, in the presence of a testis, the Müllerian system involutes under the influence of AMH secreted by the Sertoli cells, while the Wolffian system is stabilized by testosterone secreted by the Leydig cells, which are stimulated by placental human chorionic gonadotrophin (hCG), and develop into the epididymis, vas deferens, seminal vesicles, and ejaculatory ducts (Figure 7.1). It is important to note that AMH exerts its effect in a paracrine fashion (i.e. a local effect near its site of production), while testosterone acts both locally (to stabilize the Wolffian ducts) and in a classical endocrine way (to masculinize the urogenital sinus and the external genitalia) after being transported through the bloodstream to its target tissues. In the absence of a testis, the Wolffian system involutes and the Müllerian system develops into the Fallopian tubes, uterus, and the upper third of the vagina. Figure 7.1 Differentiation of the internal genitalia. Under the influence of testosterone, the sinus narrows to form the posterior urethra, and the prostate and the glands of Cowper develop from outgrowths of the sinus. In the absence of testosterone, the sinus forms the lower two‐thirds of the vagina and the urethra, and its outgrowths form the para‐urethral glands of Skene and the vestibular glands of Bartholin that secrete mucus to lubricate the vagina. From eight weeks onwards in the male, under the influence of dihydrotestosterone (DHT), converted from testosterone in genital skin by 5‐alpha reductase type 2, the genital tubercle will grow to form the penis, the urethral folds will develop into the corpus spongiosum surrounding the urethra and the genital folds will fuse in the midline to form the scrotum and ventral part of the penis (Figure 7.2). In the female, the same structures will form the clitoris, the labia minora, and the labia majora. Figure 7.2 Differentiation of the external genitalia from the common anlage. Historically, individuals with ambiguous genitalia had been classified according to the histology of their gonads. Thus, those with both ovarian and testicular tissue were called true hermaphrodites (now ovotesticular DSD), those with only ovarian tissue female pseudohermaphrodites and those with only testicular tissue male pseudohermaphrodites. Since the discovery of the human sex chromosomes in 1956, gonadal biopsies are only required to establish a diagnosis of ovotesticular DSD, a very rare occurrence. Table 7.1 shows the nomenclature proposed in 2006. Of note, mixed gonadal dysgenesis (most often associated with 45,X/46,XY mosaicism), the second most common cause of ambiguous genitalia (after virilizing congenital adrenal hyperplasia [CAH]), occupies a separate niche in both the old and the new nomenclatures. Table 7.1 Previous and new classification of disorders of sex development. By far the commonest form of 46,XX DSD is CAH from 21‐hydroxylase deficiency. The pathophysiology of this condition and its medical management are described in detail in Chapter 8. In genetic females affected with the severe form of the disease, masculinization of the external genitalia is usually detected at birth. However, virilization can sometimes be relatively mild and may be missed in the neonatal period. At the extreme end of the spectrum, masculinization may be complete, with a penile urethra, and the neonate may be thought to be a boy with bilateral cryptorchidism. Figure 7.3 illustrates the spectrum of virilization of the external genitalia that can be observed in 46,XX infants with CAH due to 21‐hydroxylase deficiency. Figure 7.3 The spectrum of virilization of the external genitalia that can be observed in genetic females with CAH due to 21‐hydroxylase deficiency. Much less common enzyme defects that can cause in utero virilization of 46,XX foetuses involve 11‐β‐hydroxylase and aromatase deficiencies. By contrast, 3‐β‐ol‐dehydrogenase, 17‐α‐hydroxylase and P450‐oxidoreductase (POR) do not typically induce in utero virilization of 46,XX foetuses. Synthetic defects in the adreno‐gonadal enzyme pathways from cholesterol to testosterone (and in its reduction to DHT in genital skin) can cause ambiguous genitalia in 46,XY individuals in spite of the presence of two normally differentiated testes (Figure 7.4). All of these enzyme defects are inherited in an autosomal recessive fashion. Specifically, 3‐β‐ol‐dehydrogenase and 17‐β‐hydroxysteroid dehydrogenase deficiencies will result in reduced testosterone production during the embryonic period and therefore incomplete virilization. 5‐α‐reductase deficiency, which results in decreased DHT production in genital skin, should also be considered in incompletely virilized 46,XY neonates. Lastly, a deficiency of the steroidogenic acute regulatory (StAR) protein, which transports cholesterol inside the mitochondria (see Chapter 8 on adrenals), also causes an autosomal recessive form of 46,XY DSD, generally with a completely female external appearance and severe adrenal insufficiency. Likewise, the under‐virilization of 46,XY newborns with Smith–Lemli–Opitz syndrome, which is due to a deficiency of 7‐dehydrocholesterol reductase and is associated with multiple congenital malformation, can be very severe. Figure 7.4 Pathways of steroid synthesis in adrenal gland and gonads. Those specific to the adrenals are found on Figure 8.2. Leydig cell hypoplasia is suggested by severe under‐masculinization, occasionally with female external genitalia, in a genetic male with identifiable gonads and raised gonadotrophins. Many individuals with this condition have been shown to have biallelic inactivation of the LH (luteinizing hormone)/hCG receptor. In contrast, because foetal pituitary LH only begins to control testosterone production by Leydig cells after 13 weeks of gestation, congenital gonadotrophin deficiency typically presents with micropenis and cryptorchidism, but not with abnormal sex differentiation. Testicular dysgenesis may also be associated with heterozygous inactivating mutations in steroidogenic factor 1 (SF‐1); adrenal insufficiency may be associated. In 46,XX subjects SF‐1 mutations are associated with premature ovarian failure. However, a specific diagnosis remains elusive in many under‐virilized males with testicular dysgenesis. Complete androgen insensitivity syndrome (CAIS) is an X‐linked recessive disorder caused by inactivating androgen receptor (AR) mutations. These mutations can occur de novo or be inherited through the mother who is a healthy carrier. There may be a history of amenorrhea and infertility on the maternal side of the family. Half of the genetically male offspring of carrier mothers will be phenotypic females with absent Müllerian structures, intra‐abdominal or inguinal testes, and a short vagina. The diagnosis is usually made in the index case upon the discovery of gonads on palpation or at operation for an inguinal hernia in a female infant. With the increased use of amniocentesis, the diagnosis may also be suspected when a phenotypically female baby is born after a 46,XY karyotype has been found on amniocentesis. The ‘classical’ presentation with primary amenorrhoea in an adolescent with normal breast development but without sexual hair has become the exception. Although completely insensitive to androgen, individuals with CAIS are normally sensitive to oestradiol which is aromatized from the high testosterone levels produced at puberty. This accounts for normal breast development and female body habitus, but there is little or no pubic and axillary hair because of the androgen resistance. AMH production by the foetal testes being normal, Müllerian structures are absent, hence the amenorrhea. Mixed gonadal dysgenesis is the term applied to 45,X/46,XY mosaicism associated with ambiguous genitalia, a streak gonad on one side, and a dysgenetic testis on the other. The external and internal phenotype will depend on the degree of testicular dysgenesis. With severe androgen and AMH deficiency, the external genitalia will be under‐masculinized and both vagina and uterus will be present. Regression of the uterine horn and of the Fallopian tube on the side where testicular differentiation occurred may be seen, illustrating the paracrine action of AMH. If testicular dysgenesis is relatively mild, Müllerian regression will have occurred and the external genitalia may be male. Indeed, 45,X/46,XY mosaicism may be found in boys with unexplained short stature and features of Turner syndrome, suggesting that in these subjects the gonads are 46,XY and the growth plates 45,X. In pure gonadal dysgenesis, the gonads are reduced to fibrous streaks, the external genitalia are female, and Müllerian structures (i.e. female internal genitalia) are present. It can occur either sporadically or in familial form. This phenomenon has been found in some cases to be due to deletion or mutation of SRY. The streak gonads should be removed as soon as the diagnosis is established because of the risk of gonadoblastoma. Pure gonadal dysgenesis can also be seen in association with a 46,XX karyotype; mutations that completely inactivate the follicle‐stimulating hormone (FSH) receptor have been found in some cases, although these more commonly lead to premature ovarian failure. External genitalia that are sufficiently ambiguous to pose the dilemma of sex assignment are observed in 1 in 5000–10 000 newborns. Both parents should be seen as soon as possible. The key points in counselling are the following: Of note, premature girls, because of the lack of subcutaneous fat, may appear to have clitoral hypertrophy. In these cases, it is useful to estimate the ratio of the distance between the anus and the posterior labial fourchette to the distance between the anus and the base of the clitoris: a ratio less than 0.5, regardless of gestational age, reasonably excludes androgenization during the first trimester. The following professionals should be informed as soon as possible: If access to the professionals listed above is difficult, then transfer to a specialist centre for specific tests or for general management may be necessary. The following investigations should be carried out as quickly as possible: Figure 7.5 shows a simplified flow diagram giving the main diagnostic possibilities. As stated above, the commonest cause of ambiguous genitalia is CAH due to 21‐hydroxylase deficiency in a 46,XX newborn. The diagnosis of 21‐hydroxylase deficiency is based on the measurement of plasma 17‐OHP, with the sample being taken on day 3 (the overlap between affected and normal being too great prior to this) and the result being requested urgently. In addition, the plasma levels of androstenedione, testosterone, and dehydroepiandrosterone‐sulfate (DHEAS) will be elevated in CAH. Blood glucose should be measured, although significant hypoglycaemia is seldom present. However, babies with CAH are at risk of salt wasting and plasma electrolytes are often measured early after birth in newborns with genital ambiguity. This may give a false sense of reassurance, because plasma electrolytes are usually normal initially. Due to renal immaturity, the initial salt‐wasting episode typically occurs only in the second or third week of life, so that the infant’s weight should be carefully monitored even if the plasma electrolytes were normal initially. If there is significant weight loss (up to 10% weight loss is within normal limits in newborns but babies should regain their birth weight by 14 days of age) or if subsequent weight gain is insufficient, plasma electrolytes should be measured again and, if hyponatraemia develops, urine sodium should be measured to document inappropriate natriuresis. Figure 7.5 Main diagnostic possibilities in neonates with ambiguous genitalia. Based on their normal female internal genitalia and potential fertility, sex assignment is almost universally female in 46,XX infants with 21 hydroxylase deficiency. Likewise, newborns with ambiguous genitalia due to mixed gonadal dysgenesis are usually assigned female. The presence of a uterus in this condition makes assisted fertility possible. However, the gonads should be removed because of the risk of gonadoblastoma. Whether in CAH or in mixed gonadal dysgenesis, the psychosexual consequences of clitoral/vulvar surgery in infancy are controversial. Some jurisdictions now allow that no sex be assigned at birth, so that the individual may choose (or not) a male or female assignment after reaching the age of legal autonomy. Most parents are still uncomfortable about this option. From the point of view of diagnosis and of sex assignment, the most difficult cases are 46,XY infants with partial androgen insensitivity syndrome (PAIS) in whom the growth potential of the phallus at puberty is unpredictable. In contrast, 46,XY individuals with enzyme defects or testicular dysgenesis have normal sensitivity to androgens and can therefore be assigned a male sex if diagnosed early enough. The diagnosis in this group of individuals has classically been based on the measurement of precursor/product ratios of various plasma steroids before and after stimulation with hCG. There are many different protocols for hCG stimulation, the simplest being a single intramuscular (im) injection of 3000 units m−2 with measurement of plasma steroids five days later. However, several of the enzymes described above are present in various isoforms encoded by different genes (only one of which has deleterious mutations) and expressed in different organs so that these plasma ratios may be misleading. For example, mutations inactivating SRD5A2 will reduce 5‐α reductase activity in genital skin and cause 46,XY DSD; however, SRD5A1 is not mutated, so that the enzyme activity in the liver is intact and the plasma testosterone/DHT ratio may be normal. Molecular analysis of the relevant genes (targeted exome sequencing) is therefore being increasingly used to establish a diagnosis based on a limited clinical and biochemical phenotype. Another example is 46,XY babies with under‐virilization and salt wasting, but normal testes, who are likely to have 3‐β‐ol‐dehydrogenase deficiency until proven otherwise and in whom the diagnosis can be readily confirmed by sequencing HSD3B2. Ultimately, criteria for sex assignment include the specific diagnosis, presence of female internal genitalia (i.e. vagina, uterus), size and androgen sensitivity of the genital tubercle, parental wishes, and cultural considerations. The advice given as to sex assignment should ideally result from a consensus between expert professionals and the parents. In cases where parents and health care providers disagree and psychological support was not able to change the parents’ choice, it is probably wise to let the parents’ opinion prevail. Management of CAIS should start with intensive counselling of the individuals, of their parents and of all health professionals involved, to dispel any notion of ‘ambiguity’. Because intra‐abdominal testes have a high propensity to develop cancer, orchidectomy is recommended. The age at which orchidectomy should be performed is controversial: if the diagnosis of CAIS is certain, some argue that it is preferable for the individual to allow breast development from her endogenous oestrogens. Testicular tumours have been reported as early as at 14 years but remain rare. For this reason, some adult women with CAIS prefer to forego orchidectomy and to undergo ultrasound surveillance. In contrast to CAIS, PAIS results in a wide phenotypic spectrum ranging from severely ambiguous genitalia to isolated male infertility. The management of these cases is difficult and controversial unless masculinization is relatively mild, in which cases they should be treated as subjects with CAIS; however, orchidectomy should be performed before puberty to avoid the risk of virilization at that time. A greater proportion of infants with PAIS are now assigned male than in the past. Boy or girl? This is usually the first question asked in the delivery room. It is therefore quite understandable that the birth of a child with sufficient ambiguity of the external genitalia as to make immediate sex assignment impossible is associated with great discomfort on the part of the health professionals and with great parental distress. On the other hand, the discovery, perhaps many years after birth, that the child has abnormal gonads and will be unable to have children without assistance, if at all, is also deeply wounding to many parents. Whatever the time when the DSD is discovered, it is essential for the clinician to invest as much time as possible in counselling parents. It is unrealistic to expect that all the information will be assimilated by the family at the first encounter, and several sessions will be required. It is also unrealistic to rely too much on printed information (e.g. parent booklets) in the early stages. Repeated explanations, assisted by hand drawings, and tailored to the parents’ psychological state, education, and ability to comprehend require time, patience, and experience. In addition to hormonal and surgical treatments as appropriate, these individuals and their parents need long‐term access to mental health professionals knowledgeable about DSDs. The current thinking is to inform the parents in full, including about the karyotype, at the time of diagnosis. The child should also be informed in a manner appropriate for his or her stage of cognitive and emotional development. Examination of the genitalia by staff and trainees at follow‐up visits should be restricted to what is strictly necessary. If the child is properly counselled by the paediatrician and parents during the prepubertal years, it will be possible to give a full and honest account of the problem at adolescence. The classically difficult scenario is explaining the condition to a girl with CAIS. It may be helpful to phrase the explanation as follows: The vast majority of girls and women have two X chromosomes and ovaries but some otherwise normal women have XY chromosomes and testes in the abdomen instead of ovaries; yet, they are completely resistant to testosterone, which is converted to oestrogen, resulting in normal breast development. However, there is no uterus and therefore there will be no menses and pregnancy would require a uterine transplant, which is currently experimental. Communication with women with the same condition is often helpful. Notwithstanding the use of the approach outlined above from diagnosis onwards, feelings of secrecy and shame remain common. These should be addressed so that by the time of transfer to adult care, the person can explain his or her condition and have as little embarrassment about it as possible. At the end of puberty and after full disclosure, people with DSDs should be transferred to an endocrinologist or a gynaecologist with expertise in these disorders. How this is organized in practice will vary according to local circumstances, but ensuring that the transfer has actually happened is important. Especially in individuals who do not need chronic medication, it is not unusual that they do not show up at the appointment made with the physician in charge of overseeing their evolution and management in adult life. Even individuals on chronic replacement therapy may stop taking their glucocorticoids and present with acute adrenal insufficiency, while gonadectomized or hypogonadal individuals who stop their sex hormones are at risk of developing osteoporosis at an early age. Studies of subjects with DSD in adulthood are few. The best studied, most numerous and relatively homogeneous group is that of women with CAH. Clearly, the surgical techniques used in the past for feminizing genitoplasty have led to sexual difficulties in many women. When adherence to treatment is good, the clitoral hypertrophy should not progress after birth and clitoral surgery can be avoided. Separating the urethral and vaginal orifices is still performed based on the rationale that a urogenital sinus is a risk factor for ascending urinary infections but even that rationale is questionable. People with 46,XY DSDs born with ambiguous genitalia are much more heterogeneous. Reports of female‐to‐male self‐reassignment at adolescence of 46,XY subjects with 5‐α reductase deficiency whose testes had not been removed in infancy are not generalizable. Importantly, almost half of 46,XY DSD subjects (mostly PAIS), reared male or female, reported that they were neither well informed about their medical and surgical history nor satisfied with their knowledge. This emphasizes the importance of the philosophy of full disclosure that is outlined above and that should now be adopted by all interdisciplinary teams following individuals with DSD. Gender non‐conformity in individuals with typical male of female genitalia has received considerable attention in recent years, both among health professionals and in society at large. This complex and rapidly moving field cannot be reviewed here. We think DSD, sexual orientation, and gender non‐conformity are best conceptualized as distinct entities. However, being born with atypical genitalia (and/or androgen exposure of the brain in 46,XX foetuses with CAH) has long‐lasting consequences. In girls with CAH, stereotypical male toy preference has been well documented in childhood; homosexual orientation and gender non‐conformity in adulthood appear to be substantially increased. This benign problem in infant girls is likely due to their low oestrogen milieu. Indeed, twice daily application of oestrogen‐containing creams for one to two weeks usually relieves the problem. Longer periods of application of oestrogen creams may lead to breast budding. Referral to a paediatric gynaecologist is only exceptionally needed. If the vaginal opening remains completely obstructed in spite of treatment with topical oestrogen, a pelvic ultrasound will rule out the Mayer‐Rokitansky‐Küster‐Hauser syndrome (congenital absence of the vagina and uterus). However, to avoid undue anxiety, this should be performed by a radiologist with paediatric expertise, given the small size of the normal prepubertal uterus. As described above, this is a common finding in premature infants and tends to become less obvious with the development of subcutaneous fat. On the other hand, if it is first noted after birth but is isolated (i.e. not associated with accelerated linear growth), the probability that the girl has a ‘simple virilizing’ form of CAH (i.e. without salt wasting) is low. A short period of watchful waiting may be in order, but if clitoromegaly increases, an androgen‐producing tumour of the adrenals or ovaries should be considered. The length of the penis should be evaluated by pressing firmly on the prepubic fat and should be compared with the existing growth curves. These show that the 10th centile for penile length in term newborns is 2.5 cm. In otherwise normal newborns with true micropenis, the presence of testes should be ascertained. If normal testes are felt with certainty, isolated gonadotrophin deficiency remains possible but can be easily ruled out by measuring plasma testosterone at about two months of age; indeed, the ‘minipuberty of infancy’ creates a critical window of opportunity to assess the integrity of the hypothalamo–pituitary–testicular axis. If no testes are felt, bilateral anorchia (which results from bilateral testicular torsion some time after sex differentiation is complete) can be ruled out by measuring plasma AMH levels as early as three days after birth (when FSH may still be normal due to inhibition by maternal oestrogens). In infant boys with micropenis due to either gonadotrophin deficiency or anorchia, sensitivity to androgens is normal and treatment with testosterone enanthate (25 mg im monthly for three months) will result in a doubling of penile size. Penile length increases until about four years of age and then remains stable until puberty. Therefore, by far the most common reason for referral for ‘micropenis’ at school age is for obese boys with a penis that is buried in prepubic fat. Anosmia may suggest an associated gonadotrophin deficiency, but in its absence these boys can be reassured by a simple physical examination as described above. The best time to accurately assess the presence and position of the testes is the first year of life, before the cremasteric reflex sets in. Although testicular descent may not be complete at birth in some normal term newborns (in which case one may take advantage of the ‘minipuberty’ to exclude gonadotrophin deficiency as outlined above), the testes should have reached a scrotal position by the end of the first year. Parents should be informed that their son has two normally descended testes and this should be noted in the health record. If this is not done, undue concern will arise at a later age and sometimes lead to superfluous investigations and even unnecessary surgeries. Indeed, even by careful examination with warm hands, many school‐aged boys will have testicles that are difficult to palpate. The vast majority of these boys have retractile testes and their testes will descend permanently in the scrotum at the beginning of puberty. If true cryptorchidism is established after repeated examinations, the extent of investigations will depend on its unilateral or bilateral nature and on whether the testes are palpable or not. In the latter case, plasma AMH will establish whether there are intra‐abdominal testes (in mid‐childhood, plasma FSH may be normal even in the absence of functional gonads). How these intra‐abdominal testes can be located by imaging is beyond the scope of this chapter; suffice it to say that ultrasound examinations are useless except if the testes are in the inguinal canal. Otherwise normal boys with incompletely descended but palpable testes, either unilaterally or bilaterally, can be referred for surgery without investigation. Hypospadias is defined as incomplete fusion of the urethra with the meatus on the ventral aspect of the penis – the glans, corona, shaft of penis, and (most severe) the perineum. This abnormality is commonly accompanied by chordee – curvature of the penis – caused by fibrosis of the corpus spongiosum. Isolated hypospadias (hypospadias without micropenis and with normally descended testes in a normal scrotum) is rarely associated with an endocrine disorder. Surgeons should be asked to refer the following conditions for further investigation, including karyotype: However, even extensive investigation of such individuals does not always lead to a specific diagnosis.
Differences in Sex Development and Common Genital Anomalies
Introduction
Normal sex differentiation and its genetic and hormonal control
Basic concepts of the embryology of sex differentiation
Genetic control of gonadal sex determination
Internal genitalia
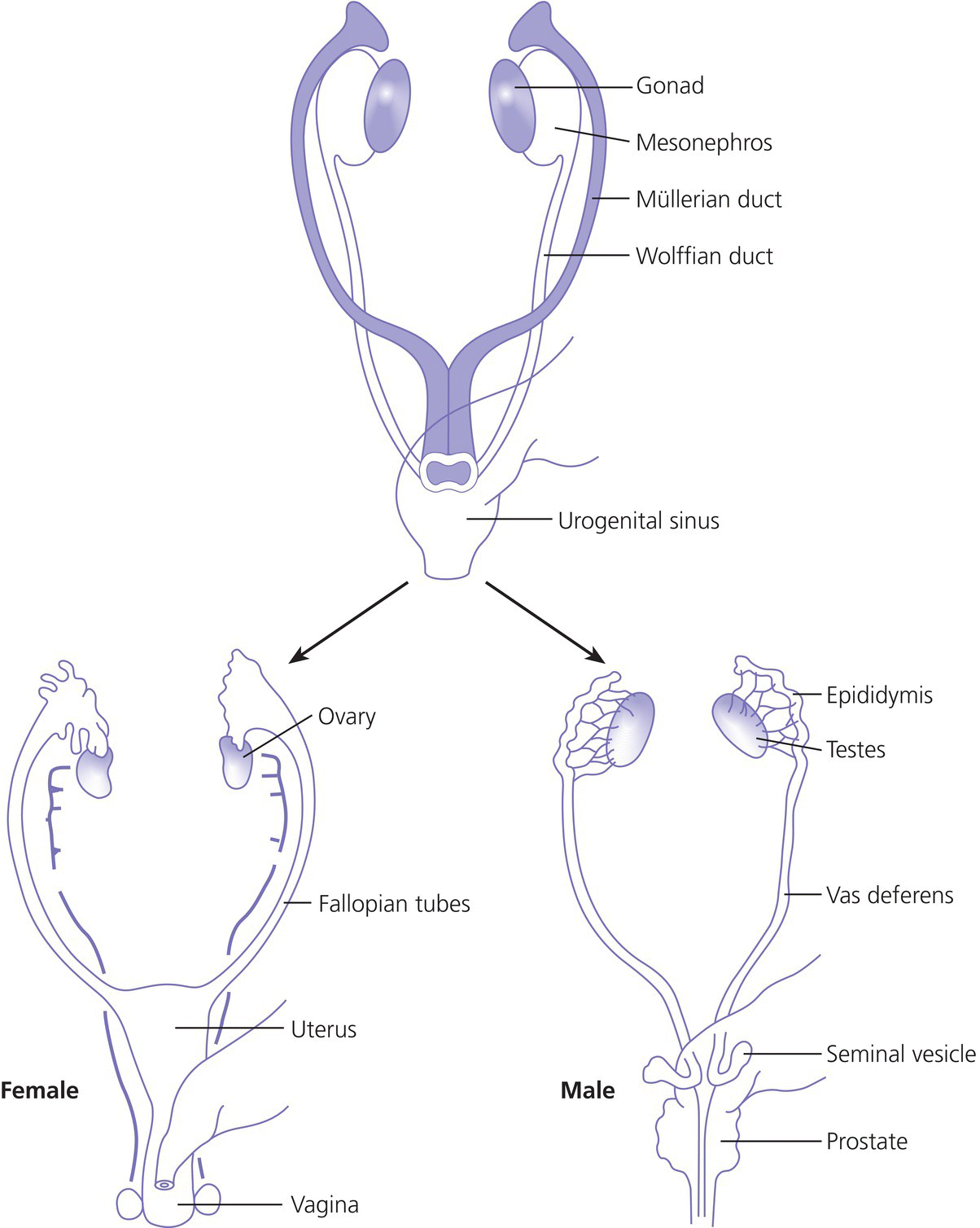
Urogenital sinus
External genitalia
Classification of DSDs
Previous
New
Intersex
Disorders of sex development (DSD)
Male pseudohermaphrodite
46, XY DSD
Undervirilization of an XY male
Undermasculinization of an XY male
Female pseudohermaphrodite
46, XX DSD
Overvirilization of an XX female
Masculinization of an XX female
True hermaphrodite
Ovotesticular DSD
XX male or XX sex reversal
46, XX testicular DSD
XY sex reversal
46, XY complete gonadal dysgenesis
46,XX DSDs
46,XY DSD
Biosynthetic defects
Abnormal testicular development (testicular dysgenesis)
Androgen resistance
Mixed gonadal dysgenesis
Pure gonadal dysgenesis
Initial investigation of DSDs
History
Examination
Further investigations and management
Aetiological diagnosis, sex assignment, and initial management of DSDs
Psychological challenges faced by individuals with DSDs and outcome
Transition to adult care
Sexual orientation and gender identity in subjects with DSD
Common genital anomalies with no ambiguity
Fused labia
Isolated clitoromegaly
Micropenis
Cryptorchidism
Hypospadias
Future developments
Potential pitfalls
Controversial points
When to involve a specialist centre
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


