Fig. 3.1
The relationship between yield, number of steps, and type of fungal template preparation method. The greatest yield comes from laborious methods that require numerous steps, including organic extractions. In contrast, the easiest methods yield the least DNA, which is typically lower in quality and more likely to be contaminated with cellular material
An unfortunate characteristic regarding fungal template preparation is that templates can be from a variety of sources, not just pure culture. The advantage of having a live culture is that there is a limitless supply of starting material for template preparation because any problems can be addressed by simply subculturing and starting over. However, the absence of a pure culture can be quite common in fungal infections, which, in fact, can increase the value of any molecular assay if pure cultures are optional. Instead, starting material for template preparation can be any type of patient specimen, including blood, body fluids, whole tissue, scrapings, washes, etc. Each specimen type can bring its own problems regarding template preparation. Components in blood specimens, such as hemoglobin and lactoferrin, as well as the preservatives or anticoagulatives in specimen collection, such as ethylenediaminetetraacetic acid (EDTA) and heparin, can inhibit downstream steps such as PCR, if they are not removed during template preparation [10]. Tissue specimens that are only lightly colonized with an infecting fungus can swamp template preparations with host nucleic acids, proteins, etc. such that fungal material cannot be purified or cannot be detected against the excess background of host material. Furthermore, tissue specimens too often can be limited in amount, preventing multiple preparations. Alternatively, depending on the source, specimens from non-sterile sites such as trauma, sputum, or bronchial lavage can be contaminated with other fungi that are not invasive, but, nonetheless, could show up in assays that are pan-fungal in nature [11, 12]. Finally, there is the problem of formalin-fixed paraffin-embedded (FFPE) tissue specimens, which confer difficult challenges in spite of the wide availability of commercial kits that claim to be effective for processing these types of specimens. These specimens often make molecular detection difficult because nucleic acids can be cross-linked or fragmented during the fixation, leading to non-amplification by PCR [13]. They also can be externally contaminated [14], in addition to the ever-present problem of efficiently breaking the fungal cell walls once they have been suitably deparaffinized. Unfortunately, fixed specimens are among the most valuable fungal diagnostic specimens, so there is a substantial opportunity for template preparation improvement.
As trivial as template preparation seems, this step may be the most important area of molecular diagnosis to develop and standardize. Unfortunately, because of the diverse nature of fungal cell walls, with yeasts generally being easier to lyse than molds, it has been difficult to devise a “one size fits all” method. Until this challenge is overcome, new technologies will not be able to assume most of the diagnostic burden in a clinical laboratory.
Diagnostic Strategies
Application of molecular biology to fungal diagnosis has drawn from many areas, which has resulted in the successful use and continued development of numerous molecular platforms for fungal identification. The stringent conditions that clinical microbiology laboratories operate under combined with the multiple isolate sources (pure culture, human specimens, fixed tissue, etc.) pose difficult challenges to the widespread use of molecular biology in clinical mycology. However, the numerous working assays and the continual development of new technologies offer great promise for one or more platforms to become standard equipment in a clinical laboratory (Table 3.1).
Table 3.1
Comparison of different diagnostic platforms
Method | Capital cost | Technical expertise | Assay cost | Turnaround time | Downstream analysis | Labor | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|
PCR | Low | Lowest | Low | Low | Low | Low | High | High |
qPCR | Medium | Low | Low | Low | Low | Low | Highest | Highest |
NASBA | Lowest | Lowest | Low | Low | Low | Low | High | High |
LAMP | Lowest | Lowest | Low | Low | Low | Low | High | High |
RCA | Lowest | Lowest | Low | Low | Low | Low | Highest | Highest |
REP-PCR | Low | Low | Low | Medium | Medium | Low | High | Medium |
RAPD | Low | Lowest | Low | Medium | Low | Medium | Lowest | Lowest |
Sequencing | Highest | Highest | Low | Medium | Medium | Medium | High | High |
Array | High | Low | High | Medium | Low | Low | High | High |
PNA-FISH | Medium | Low | Low | Low | Lowest | Lowest | High | Highest |
MALDI-TOF | Highest | High | Lowest | Lowest | Lowest | Lowest | Medium | High |
Polymerase Chain Reaction (PCR)
Among the most basic of all molecular assays, and the most common molecular technique used by mycologists, is PCR. This technique is roughly 30 years old and is the workhorse of molecular biology, with almost limitless uses [15]. There are countless diagnostic uses and, importantly, an extensive array of variations of the basic technique that have specific applications.
Conventional PCR
The most basic PCR strategy is conventional PCR, which consists of two primers that anneal to complimentary target sequences. Taq polymerase, in addition to the appropriate buffer mix and template DNA, is used with a thermocycler to yield a product from a suitable starting template. This product can be the end point of the assay when visualized on an ethidium bromide gel, or it can be digested to yield specific restriction patterns that can be informative. Typically, conventional PCR assays have as an end point the presence or absence of an amplification product and almost always need to utilize specific primers that typically amplify a single or very narrow range of fungi. Size markers are used to confirm that the product is from the correct target since the amplicon size is usually known based on primer position. While degeneracy or conserved target sequences can extend the species target range, this extension can defeat the purpose if the goal is species identification. In some cases, the goal can be detecting the presence of a specific gene or allele, which could carry pathogenic or drug susceptibility consequences when it is present. Continued research has resulted in many variations that have made the technique more powerful, more sensitive, cheaper, and more creative in its applications.
Quantitative (Real-Time) PCR
The most common platform of nonconventional PCR for fungal detection is quantitative real-time PCR (qPCR), which offers a number of advantages over the older gel-based conventional PCR approach. The major advantage of qPCR is that it can be used to quantitate fungal colony-forming units, or the equivalent, and it is generally faster since the technique incorporates a target-specific, fluorescently labeled probe, or a general nonspecific dye that preferentially binds double-stranded DNA. The small amplicon size in qPCR, sometimes less than 100 bp, greatly decreases cycling time as amplicons can be one tenth the size of conventional PCR amplicons. Labeled probes confer the extreme specificity that qPCR is known for, although they add substantially to reaction costs compared to a nonspecific DNA-binding dye such as SYBR Green. However, suppliers of qPCR probes often allow web-based, specific assay design using algorithms that precisely match the two PCR primers and probe in a single reaction, which can be purchased as an individual assay, often as a ready-to-go PCR kit complete with enzyme and other reaction components. The programs work on a user-supplied target sequence that is simply pasted into the website to yield each assay with individual components tailored to the most efficient amplification of the target. These programs can be proprietary, available on the Internet, or part of desktop molecular biology programs.
Reactions are performed in a dedicated thermocycling instrument that can detect multiple dyes that fluoresce at different wavelengths, which can allow multiplexing in a single tube. There are various classes of fluorescent dye technologies for target detection, depending on the need. Although a Food and Drug Administration (FDA)-approved fungal diagnostic assay using qPCR technology has not reached clinical laboratories, qPCR is used extensively for the detection and quantitation of other microbes; consequently, it is only a matter of time before a specific fungal assay is routinely used in the clinical laboratory. However, it has been applied to virtually all of the major and more common human fungal pathogens and is frequently a part of most research laboratories.
Nucleic Acid Sequence-Based Amplification
There are a number of conventional PCR variations that have displayed clear value in the area of diagnostics. Some of them are moving away from the thermocycler platform and into isothermal amplification, which has the major advantage of greatly reducing instrumentation costs because reactions are run at a single temperature that can be reached with a heating block. Nucleic acid sequence-based amplification (NASBA) is a constant temperature (41 ℃) transcription-based amplification method that uses RNA as a template and a combination of RNase H and T7 RNA polymerase as enzymes. There are numerous advantages of this technique as it amplifies RNA templates, which can be present in great abundance for some targets (i.e., rRNA). Because RNA is the template, exogenous amplification of contaminating DNA is eliminated. The constant temperature and single-tube reaction also reduce contamination possibilities. A commercial version of this technology (AccuProbe, Hologic Gen-Probe Inc., San Diego, CA) has been available for a number of years and can be used to detect Histoplasma capsulatum, Coccidioides immitis, and Blastomyces dematitidis [16–18].
Loop-Mediated Isothermal Amplification
A second type of isothermal PCR is called loop-mediated isothermal amplification (LAMP) [19]. The key component of this reaction is a DNA polymerase derived from the bacterium, Bacillus stearothermophilus. Reactions can be completed within an hour and are performed isothermally at a temperature range of 60–65 ℃. In addition to the isothermal temperature and relatively fast turnaround time, LAMP is resistant to exogenous contaminants due, in part, to the requirement for four primers. Therefore, template nucleic acids do not need to be highly purified, allowing cruder, “dirty” preparations to be used; however, the large number of primers can add complexity to primer design compared to traditional PCR reactions that utilize two primers [20]. There are multiple product detection methods, which vary from ethidium bromide-stained gels to turbidity, and even, to fluorescence. LAMP has been successfully used to detect a number of fungi, including Fusarium spp., Cryptococcus spp., and Aspergillus spp. [21–24].
Rolling Circle Amplification
Rolling circle amplification (RCA) is a third type of isothermal amplification technology that can result in the massive amplification of target sequences (~ 109 fold) [25]. This technology utilizes bacteriophage Phi29 DNA polymerase to amplify a DNA template. RCA is highly sensitive and specific, and can be used to discriminate single nucleotide polymorphisms. The technology utilizes a circular template to generate linear amplification products, which can be detected fluorescently using a variety of different probe chemistries. This sensitivity is particularly important for fungal genera that can be difficult to distinguish at the species level, even with molecular methods, due to highly conserved target sites [26]. The method is generally refractory to inhibitory compounds present in a sample that would normally inhibit other types of PCR reactions. RCA can also be easily multiplexed in contrast to conventional PCR so that multiple targets can be detected simultaneously. RCA has been used to detect a variety of fungi, including Penicillium marneffei, Fonsecaea spp., and Exophiala spp. [27–29].
Random Amplification of Polymorphic DNA
In addition to these technologies, there are various other PCR-based methods that have various applications in diagnostic mycology. For example, PCR can be used for some aspects of epidemiology. For fungi, this application can be extremely important because in many cases, little is known about the infecting fungus. A major PCR-based application in fungal epidemiology is random amplification of polymorphic DNA (RAPD). RAPD is a fingerprinting method that utilizes single, short oligonucleotides (~ 10 bp) in a PCR reaction typically at low annealing temperatures. PCR products can be produced when two oligonucleotides anneal close enough together on complementing DNA strands to produce a PCR product. Running the PCR reaction on an agarose gel followed by staining reveals an isolate-specific pattern that can be used to distinguish two isolates from one another, depending on the oligonucleotide. The discriminating power of the assay relies on multiple products being produced in a single reaction. Genetically unrelated strains are predicted to have different RAPD patterns depending on how fast their genomes evolve, geographic isolation from each other, and the frequency of the primer site in the genome. The greatest advantage of RAPD as an epidemiological tool is that nothing needs to be known about the genome sequence of the organism. The primer choice can be made empirically by testing single primers on one isolate and selecting the primer that yields the most bands for further testing on unrelated strains. For some fungi, such as Cryptococcus neoformans, this method has been studied extensively and has been developed into a strain typing tool based on PCR pattern [30]. The method can be highly susceptible to variation, even within laboratories, and is therefore not generally used as an identification tool. However, it has tremendous utility as a rapid way to investigate outbreaks to determine if infections are from the same or unrelated strains.
Repetitive Sequence PCR
Repetitive sequence PCR (rep-PCR) is both an epidemiologic and diagnostic tool that can, in some cases, identify as well as discriminate individual fungal strains [31]. The method is PCR based and relies on the high frequency of repetitive elements found in some microbial genomes, including bacteria and fungi. Using a large collection of stock primers in a semiautomated platform, the method can be used to fingerprint fungi as well as identify some species of fungi by searching the generated fingerprint pattern against a reference library [32, 33]. The method is well advanced and has already been commercialized (DiversiLab, bioMérieux, Inc., Durham, NC).
Sequencing
While live culture is generally considered the gold standard of fungal diagnosis, arguably, the “gold ring” of molecular diagnostics should be a system that can identify any fungus, independent of any accompanying information (patient history, symptoms, imaging, morphology, biochemistry, serology results, etc.) using a single assay. While there are a number of methodologies that may be able to fulfill this possibility, the technology that is farthest along is sequence-based identification. Key factors that have enabled sequence-based identification to be as powerful as it is are the easy access to public databases, such as GenBank, and the search algorithms such as the BLAST programs [34] that enable users to query the database with an unknown sequence. Inexpensive sequencing, through advances in instrument technology and chemistry, has enabled the generation and deposit of sequences from countries all over the world, leading to both a deep and diverse database of easily searchable fungal sequences. The field of bioinformatics has made it possible to determine which sequences are the most informative and discriminatory with regard to species identification. Bioinformatics has advanced hand in hand with the completion of genome sequences, of which hundreds of fungal genomes are now complete. Next-generation sequencing will greatly accelerate genome sequencing and may someday be as fast and inexpensive as obtaining a single gene sequence.
Sequence-based identification of fungi evolved into a powerful diagnostic tool with the deposit of increasing numbers of ribosomally derived sequences (18s subunit, internal transcribed spacer regions 1 & 2, 5.8s subunit, 28s subunit, and the intergenic region). These sequences were important for fungal phylogeny and drove diagnostic mycology into the molecular era; however, as the deposits accumulated, both conserved and variable regions were identified. The existence of these regions allowed the design of primers to conserved regions, which enabled PCR amplification and sequencing of an unknown fungus through universal primers that could anneal to the conserved sites, and amplify the unknown intervening regions. The resultant amplicons contain the conserved priming sites in addition to the informative intervening regions, which enabled identification, often to the species level. Two of the most common pairs of primers are the ITS1 and ITS4 and the NL1 and NL4 primers (Fig. 3.2) [35, 36]. There are many other primers that are suitable for amplifying informative regions. However, these primers were used to deposit many of the fungal sequences into GenBank that are useful for diagnostic purposes. These two pairs amplify informative regions containing the internal transcribed spacer (ITS1 and ITS2) regions as well as the D1/D2 region of the 28s or large ribosomal subunit. Both regions are informative when searching GenBank, although the region amplified by the ITS1 and ITS4 primers is generally more variable than the region amplified by the NL1 and NL4 primers. Furthermore, the ITS region contains two individually informative regions, the ITS1 and ITS2 regions, which are separated by the 5.8s subunit. A third potential amplicon, in addition to the ITS and D1/D2 regions, is the intergenic sequence, sometimes referred to as the IGS region (Fig. 3.2). This region separates the 28s and 5s subunits and is extremely variable. However, it is generally too long for universal primers to be used in a PCR reaction, although it can be used to distinguish specific species, but has not been applied to general fungal molecular identification.
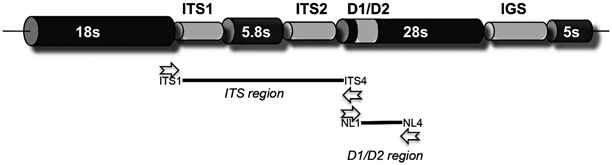
Fig. 3.2
Organization and primer location of the fungal ribosomal genes. The ITS region, generally the most informative, can be amplified by the ITS1 and ITS4 primers, and is located between the end of the 18s ribosomal subunit and the 28s ribosomal subunit. The D1/D2 region if found within the 28s ribosomal subunit towards the 5s end and can be amplified with the NL1 and NL4 primers. The IGS region is also quite variable and is typically but not always found between the 28s and 5s subunit. However, the size of this region is generally too large to amplify in a PCR reaction. Not drawn to scale. Open arrowheads indicate primer sites
In addition to the conserved and near-universal PCR priming sites, a second important factor for targeting the rDNA for molecular identification is copy number. Eukaryotic ribosomal subunits are typically present in multiple copies that can exceed single copy targets by 10–100x. This increased copy number confers greater sensitivity for PCR reactions, which can be extremely important when template is not prepared from pure culture but, instead, from tissue specimens, body fluids, or other samples that may have a low or limited amount of organisms. In addition to some of the multiple mitochondrial targets, the multicopy nature of these targets is indispensable for limited amounts of cellular material, as is often the case when patient specimens must be tested directly.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


