Diabetic Ketoacidosis
INTRODUCTION
Diabetic ketoacidosis (DKA) is an acute metabolic complication of diabetes characterized by an absolute or relative insulin deficiency resulting in hyperglycemia, hyperketonemia, and metabolic acidosis. DKA is an endocrine emergency that requires immediate treatment and close monitoring for metabolic derangements. It most commonly occurs in patients with a known history of type 1 diabetes; however, patients with type 2 diabetes are also at risk during states of acute illness. In some cases, DKA may be the first manifestation of diabetes, especially in developing countries.
DEFINITION
The diagnostic criteria for DKA include hyperglycemia with a blood glucose level greater than 250 mg/dL, the presence of serum ketones, an anion gap metabolic acidosis with a pH less than 7.3, a serum bicarbonate less than 18 mEq/L, and an anion gap greater than 12.
Euglycemic Diabetic Ketoacidosis
Euglycemic DKA (euDKA) is characterized by an anion gap metabolic acidosis and hyperketonemia as in traditionally described DKA, but with the exception that patients present with a serum glucose level less than 200 mg/dL.
EPIDEMIOLOGY
DKA continues to be an important cause of morbidity and mortality among patients with diabetes. The prevalence of DKA hospitalizations has been on the rise in the United States with an increase in the rate from 19.5 to 30.2 per 1000 persons with diabetes between 2009 and 2014. This increase in prevalence was consistent across all age groups and sexes, with the highest rates seen in persons aged less than 45 years. The reason for the rise in hospitalization rates for DKA is unknown. Possible explanations include the rising incidence of type 1 and type 2 diabetes in children and adolescents, suggesting that more patients are presenting with DKA at diagnosis. There is also an increase in provider awareness and identification of milder forms of DKA that may have previously gone unrecognized. The rates of in-hospital case-fatalities have decreased from 1.1% to 0.4% during this same time period, with the highest case-fatality rates among the elderly and patients with concomitant life-threatening conditions. Patients presenting with shock or coma on admission had a worse prognosis, with the main causes of death being infection, hypokalemia, and circulatory collapse. DKA also remains as the most common cause of death in children and adolescents with type 1 diabetes.
DKA is more common among patients with type 1 diabetes, but it can also occur in patients with type 2 diabetes. Patients with poorly controlled type 2 diabetes with concurrent illnesses and patients with a history of ketosis-prone type 2 diabetes are at higher risk. Ethnic minorities, specifically patients of African-American or Hispanic descent, have a higher prevalence of ketosis-prone type 2 diabetes and typically have a strong family history of diabetes with associated negative autoimmune markers. DKA as an initial presentation for diabetes is also common, but incidence rates vary drastically between countries, ranging from 13% to 80%, with the highest incidence rates seen in developing countries.
EuDKA was first described in a case series in 1973 when 37 of 211 patients admitted for DKA were found to have mild hyperglycemia with metabolic acidosis. The majority of patients described in the case series had type 1 diabetes with associated recent carbohydrate reduction, vomiting, or decreased insulin doses. Although previously rare, there is also a growing rise of euDKA in association with sodium-glucose cotransporter 2 (SGLT-2) inhibitors. In patients with type 1 diabetes and SGLT-2 inhibitor use, the incidence of DKA has been reported to be 9.4%, as compared with 0.2% in patients with type 2 diabetes on SGLT-2 inhibitors. , In summary, euDKA is more likely to occur in patients with type 1 diabetes but can also occur in patients with type 2 diabetes.
PATHOPHYSIOLOGY
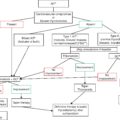
DKA is triggered by an insulin-deficient state that leads to activation of counterregulatory hormones that promote lipolysis, glycogenolysis, and gluconeogenesis, and clinically manifests with hyperglycemia, ketosis, and metabolic acidosis with profound volume depletion.
Insulin Deficiency and Counterregulatory Hormones
DKA results from an absolute or relative insulin-deficient state. Patients may become insulin deficient in a variety of settings including new-onset diabetes, nonadherence to insulin treatment regimen, insulin pump failure, medication or illicit drug use interactions, or during acute physiologic states that require an increase in circulating insulin levels. Insulin deficiency leads to a perceived fasting or low glucose state at the cellular level activating insulin counterregulatory hormones including glucagon, cortisol, growth hormone, and catecholamines. The counterregulatory hormones antagonize the effects of insulin by increasing the blood glucose level through the activation of alternative mechanisms of energy production. Glucagon plays a key role by promoting glycogenolysis, gluconeogenesis, and lipolysis. Cortisol, catecholamines, and growth hormone also stimulate lipolysis by activating hormone-sensitive lipase in adipose tissue, leading to the release of free fatty acids and glycerol. Glycerol is then recycled in the liver as an important substrate in the gluconeogenesis pathway and further promotes the rise of blood glucose, leading to hyperglycemia.
Free Fatty Acids in Diabetic Ketoacidosis
The free fatty acids released from lipolysis are transported to the liver where they become the precursors to ketoacids and further suppress the production of insulin. Under the stimulation of glucagon, the enzyme malonyl-coenzyme A (CoA) becomes upregulated in the liver, facilitating the transport of free fatty acids into the mitochondria. Once in the mitochondria, free fatty acids are limited from entering the Krebs cycle, as the key substrate pyruvate is diverted from glycolysis to gluconeogenesis during the perceived cellular fasting state. This Krebs cycle roadblock gives rise to an accumulation of free fatty acids in the mitochondrial matrix that are then catabolized into acetyl-CoA and ketoacids B-hydroxybutyrate and acetoacetate, which can be used as energy in the body. The production of ketones leads to a ketonemia that is further maintained by reduced liver ketone clearance. At baseline, ketone bodies are acidic, but the body is able to produce extracellular and intracellular buffers to neutralize the acidity. However, in DKA, the body is unable to meet the buffering demand imposed by the ketone excess, leading to an anion gap metabolic acidosis. The metabolic acidosis is also worsened by further conversion of pyruvate into lactate in the cells.
Dehydration
The counterregulatory hormones work to promote glycogen breakdown and glucose synthesis, leading to hyperglycemia. When the glucose level in the blood reaches an approximate concentration of 225 mg/dL, glucose will start to leak into the renal tubules, creating an osmotic gradient and water diuresis. The diuresis can be quite significant and lead to volume depletion and dehydration if patients cannot sufficiently maintain their oral fluid intake.
Ketones also contributes to the diuresis. As the ketone concentration accumulates in the blood, it also collects in the urine where it is combined with sodium to form a buffer that can be excreted. The creation of the sodium gradient drives further movement of water that is excreted, leading to more dehydration.
The hyperglycemia-induced osmotic diuresis also promotes the loss of other electrolytes in the urine including potassium, calcium, phosphorous, and magnesium. The fluid loss also leads to a decrease in glomerular filtration in the kidneys, which then causes a decrease in glucosuria and worsens the hyperglycemia.
Pathophysiology of Euglycemic Diabetic Ketoacidosis
The pathophysiology of euDKA is not entirely known; possible causes could be as simple as recent insulin administration before presentation and therefore masking hyperglycemia, but can also be a result of significant carbohydrate restriction, excess alcohol consumption, chronic liver disease or liver cirrhosis, glycogen storage disorders, pancreatitis, or pregnancy.
One possible mechanism in euDKA patients stems from decreased hepatic glucose production during a fasting state. Through fasting or perceived starvation, which can occur from decreased carbohydrate caloric intake, acute illness, pregnancy, or substance misuse, the body is depleted of glycogen. This leads to a milder hyperglycemia when triggered in DKA. Another proposed explanation is that some patients may have an enhanced excretion of urinary glucose caused by a surplus in counterregulatory hormones or SGLT-2 inhibitor use, leading to reduced serum glucose.
In the United States, SGLT-2 inhibitors were Food and Drug Administration (FDA) approved in 2013 for the treatment of type 2 diabetes, and work by promoting glucose reabsorption from the proximal renal tubule leading to a decrease in serum glucose and an increase in glucosuria. If a patient was in DKA while on an SGLT-2 inhibitor, the SGLT-2 inhibition provides an insulin-independent mechanism for lowering glucose levels through urinary glucose excretion with the end result being euDKA.
The association of euDKA with the use of SGLT-2 inhibitor medications prompted an FDA drug safety warning about the associated risk.
CLINICAL PRESENTATION
Clinical Findings
DKA is a rapidly evolving condition with symptoms typically presenting within a 24-hour period. Patients in DKA most commonly present with symptoms of hyperglycemia, including polyuria and polydipsia. EuDKA can pose a diagnostic challenge to providers given the presence of a milder hyperglycemia, which may lead to a delay in treatment and medical complications.
Patients may also have feelings of fatigue, weight loss, nausea, vomiting, and abdominal pain. Severe presentations of DKA may also include mental status changes that can vary from altered sensorium to lethargy and coma. On physical examination, patients may appear dehydrated with dry mucous membranes, poor skin turgor, tachycardia, and hypotension. The physical examination may also reveal a fruity breath odor as a result of elevated serum acetone levels and rapid and shallow Kussmaul breaths as a compensatory mechanism for metabolic acidosis. Features of the precipitating trigger for DKA may also be present.
Laboratory Findings
Once the clinical suspicion for DKA is established, the diagnosis must be confirmed with laboratory testing. This includes measurement of glucose, metabolic panel, pH, and serum ketones.
An immediate point-of-care fingerstick should be done to confirm hyperglycemia of greater than 250 mg/dL. It is important to note that in cases of euDKA, the blood glucose value will be less than 200 mg/dL, but the laboratory findings of an anion gap metabolic acidosis with positive serum ketones will still be present.
To assess for metabolic acidosis, an arterial sample to measure the pH along with a metabolic panel to calculate an anion gap should be obtained. If an arterial sample is not available, venous blood gas can be used to assess the pH and has a 97.8% sensitivity and 100% specificity in the diagnosis of DKA. The anion gap is calculated by measuring the difference of cations (which is represented as corrected serum sodium) and anions (which is represented as the sum of serum chloride and bicarbonate).
For ketone analysis, the American Diabetes Association recommends serum ketone testing in favor of urinalysis ketone testing to allow for a more accurate diagnosis. Ketone urinalysis only measures acetone and acetoacetate, and not B-hydroxybutyrate, which is produced in a 20:1 ratio in DKA, making it the primary ketoacid. Serum B-hydroxybutyrate testing has a sensitivity of 98% and a specificity of 79%, compared with ketone urinalysis testing, which has a sensitivity of 98% and specificity of 35%.
In addition to diagnostic testing, laboratory studies should include measurements of electrolytes potassium; magnesium, which may be low and require supplementation; and phosphate, which may be normal or initially elevated at presentation. Patients should also have a complete blood count, urinalysis, an electrocardiogram, and their hemoglobin A1c measured. Focused testing of precipitating triggers may also be indicated depending on the clinical presentation.
DIFFERENTIAL DIAGNOSIS
Many medical conditions share the individual components that together define DKA: hyperglycemia, anion gap metabolic acidosis, and ketosis. Hyperglycemia can be a marker of a variety of conditions including uncontrolled type 1 or type 2 diabetes, physiologic stress, infection, and, in severe cases, hyperosmolar hyperglycemic state (HHS). HHS presents with marked hyperglycemia, typically with blood glucose greater than 600 mg/dL. However, unlike DKA, patients with HHS will have minimal acidosis with pH greater than 7.3 and serum bicarbonate greater than 15 mmol/L, absent to mild ketosis, and elevations in serum osmolality to more than 320 mOsm/L.
Several conditions can also present with an anion gap metabolic acidosis. This list includes alcohol intoxication, uremia, rhabdomyolysis, and toxicity from salicylates, paraldehyde, methanol, or ethylene glycol ingestion. Lactic acidosis also leads to an anion gap metabolic acidosis and has its own differential including infection, pancreatitis, ischemia, and seizure.
Ketosis can occur in states of starvation. When the body is depleted of glucose, the liver will start to produce ketone bodies in order to generate ATP for the brain. Patients with a history of chronic alcoholism can also develop an alcoholic ketoacidosis. Ethanol can be metabolized to ketones but it can also suppress gluconeogenesis and lead to a perceived state of starvation.
Given this broad differential diagnosis which can have similar laboratory findings to DKA, it is important to consider the combination of laboratory diagnostics along with the clinical history and physical examination to accurately diagnose DKA.
DIABETIC KETOACIDOSIS PRECIPITANTS
DKA most commonly occurs among patients with a known history of diabetes who acutely run out of insulin or stop taking their insulin. In this scenario, it is important to investigate the barriers or causes contributing to insulin nonadherence. In addition to medication nonadherence, it is also important to consider that other precipitants and stressors which can activate an increase in the counterregulatory hormones cortisol, catecholamines, and glucagon can precipitate DKA.
Illness can be a major stressor, particularly if secondary to infection, myocardial infarction, and stroke. Illness associated with vomiting is also a common precipitant for DKA by promoting dehydration and may lead to patients omitting or decreasing the amount of insulin they are administering in order to avoid hypoglycemia.
Medications can also precipitate DKA. Corticosteroids, high doses of thiazide diuretics, atypical antipsychotics, and diazoxide have all been associated triggers for DKA. The use of SGLT-2 inhibitors has also been associated with precipitating euDKA. Illicit drug use and alcohol also increases the risk of DKA by interfering with medication adherence. More obscure precipitant causes of DKA include Cushing’s syndrome, acromegaly, glucagonoma, pancreatic destruction caused by a virus or neoplasm, and pregnancy.
TREATMENT
The management aims in DKA are to restore fluid, electrolyte, and hormonal homeostasis while minimizing the risks for complications. Treatment requires close monitoring to assess the response and resolution of hyperglycemia and to correct laboratory abnormalities. Due to the need for close monitoring, patients with DKA are best managed in an intensive care unit (ICU) or step-down unit.
Fluid Replacement
The osmotic diuresis precipitated by hyperglycemia in DKA leads to volume depletion and dehydration. Fluid resuscitation will correct the intravascular volume depletion while enhancing renal perfusion and promoting further glucose diuresis. Additionally, fluid replacement helps to decrease the counterregulatory hormone response by restoring the intravascular volume and further decreasing glucose levels in the blood.
Patients with DKA have an estimated water deficit of about 5 to 7 L, or 100 mL/kg, which represents a 10% to 15% loss in body weight. The initial goal in fluid repletion is to expand the intravascular volume and restore hemodynamic function. In the absence of heart failure, pulmonary edema, or end-stage renal disease, isotonic saline (0.9% sodium chloride) is infused at a general rate of 500 to 1000 mL/hour or 15 to 20 mL/kg per hour for the first 2 hours. If the patient is hypotensive and in hypovolemic shock from severe dehydration, 3 or 4 L of isotonic saline may be required to restore blood pressure. To assess for successful fluid replacement, patients should be hemodynamically monitored for improvements in blood pressure, heart rate, urine output, laboratory values, and physical examination.
After correcting the intravascular depletion, the remaining goal of fluid management is to replace half of the estimated fluid deficit over the course of 12 to 24 hours. If the corrected serum sodium remains low, the normal saline infusion can be continued but in general decreased to 250 to 500 mL/hour. If the corrected serum sodium level is normal or high, indicating a remaining free water deficit, then the infusion can be changed to half-normal saline and decreased to 250 to 500 mL/hour. Once the blood glucose level drops to 250 mg/dL or below, fluids should contain dextrose 5% to 10% to prevent hypoglycemia, whereas insulin therapy is still required to decrease ketones and correct acidosis.
Electrolyte Replacement
Potassium
At presentation, patients in DKA typically present with potassium abnormalities. Most commonly, patients will present with hyperkalemia despite a total-body potassium depletion. This paradox is due to the normal action of insulin to drive potassium intracellularly and out of the serum. The insulin deficiency characteristic of DKA causes a redistribution of potassium into the serum. In addition, the metabolic acidosis in DKA further causes potassium to move out of the cell as potassium is exchanged for hydrogen. As insulin therapy and fluids are administered during treatment, serum potassium levels begin to decrease. The insulin moves the potassium back into the cells, whereas the fluid repletion can cause a dilution of the serum potassium, increasing the risk for hypokalemia. Hypokalemia must be closely monitored, prevented, and treated. To prevent hypokalemia, potassium should be repleted after potassium levels fall below 5.2 mEq/L. Typically, 20 to 30 mEq of potassium is given with each liter of intravenous fluid for a goal potassium of 4 to 5 mEq/L; however, lower doses may be required in patients with decreased renal function.
In rare cases, patients in DKA may present with hypokalemia. The osmotic diuresis may lead to significant urinary losses of potassium. If the potassium level is less than 3.3 mEq/L, potassium repletion should be given with the initial fluid resuscitation and insulin treatment should be delayed to avoid cardiac arrhythmias. Once potassium levels are greater than 3.3 mEq/L, insulin therapy can be administered.
Phosphate
As with potassium, patients in DKA generally have a total body depletion in phosphate. However, most patients will have normal or increased levels of phosphate at initial presentation. The initial hyperphosphatemia is likely secondary to the concentrated levels in the setting of intravascular volume depletion and acute renal impairment. Serum phosphate levels will decrease with insulin and fluid therapy. Studies have not shown any benefit of phosphate replacement in the management of DKA, but replacement is indicated in those with severe hypophosphatemia with serum phosphate levels less than 1 mg/dL. In the event of severe hypophosphatemia, 20 to 30 mEq of potassium phosphate can be added to the fluids to prevent complications, which include cardiac arrhythmias and respiratory compromise.
Bicarbonate
Bicarbonate therapy is typically only given in DKA for patients with serum pH levels of 7 or below. There is controversy in this recommendation, as there is no current evidence that demonstrates improved DKA outcomes with bicarbonate administration. Proponents of bicarbonate advocate that bicarbonate may help patients with severe acidosis and therefore decrease risks for cardiac and vital organ dysfunction. Opponents to bicarbonate argue that bicarbonate administration can worsen outcomes by increasing the risk for pH shifts, fluid retention, and cerebellar edema.
Insulin
Insulin is used to correct hyperglycemia by increasing peripheral glucose uptake into the cells and decreasing hepatic gluconeogenesis. Insulin therapy also directly inhibits ketoacid production and decreases the release of free fatty acids. Currently, there is no consensus on a single insulin protocol for the treatment of DKA; however, all protocols share the same goals of insulin therapy – to correct hyperglycemia and restore acid–base balance.
Intravenous insulin therapy has long been favored for the treatment of DKA due to its rapid onset of action and short half-life, giving the treating provider the ability to dynamically adjust doses based on glucose response to insulin. However, there is growing interest and use of subcutaneous insulin protocols that have the advantage of decreasing the intensity of care required and possibly avoiding the need for an ICU admission for relatively mild cases of DKA.
Insulin should only be started after initiating fluid resuscitation and correcting hypokalemia, if indicated, to avoid worsening cellular fluid and potassium shifts. When insulin therapy is started, it is important to closely monitor glucose levels every hour, in addition to monitoring serum electrolytes, glucose, magnesium, phosphorous, blood urea nitrogen, creatinine, and venous pH every 2 to 4 hours.
Most intravenous insulin protocols utilize a weight-based dose for the infusion rate (0.1 to 0.15 units/kg per hour) with titration based upon the change in glucose each hour and a goal of a decrease between 50 and 75 mg/dL per hour and a target blood glucose of less than 200 to 250 mg/dL. Some protocols may also utilize an intravenous bolus of insulin before starting an insulin infusion.
RESOLUTION OF DIABETIC KETOACIDOSIS
Resolution of ketoacidosis occurs when the blood glucose level is below 200 mg/dL, serum bicarbonate levels are greater than 18 mEq/L, venous pH is greater than 7.3, and the calculated anion gap is less than 12 mEq/L. Once these targets are reached, patients can be bridged to subcutaneous insulin therapy from intravenous insulin therapy. It is important for there to be a period of overlap during the transition to avoid iatrogenically causing an insulin-deficient state and the possibility of the patient re-developing DKA. To avoid this, the basal insulin dose should be administered and the intravenous insulin infusion can be discontinued 2 hours after the dose is given.
Patients with a known history of diabetes can typically be given their home dose of basal insulin. Patients with a new diagnosis of diabetes can be started on a weight-based insulin regimen or insulin doses can be estimated based on total insulin required during resolution of DKA.
COMPLICATIONS OF DIABETIC KETOACIDOSIS
Hypokalemia and Hypoglycemia
The most common complications in the management of DKA include hypoglycemia and hypokalemia. Hypoglycemia can occur from high insulin infusion rates without sufficient intravenous fluid dextrose administration. Although there is no universal protocol for insulin dosing, the need for treatment monitoring with monitoring of hourly glucose to both prevent and identify hypoglycemia is critical. Monitoring is especially important because some patients in DKA may not develop traditional symptoms of hypoglycemia due to a blunted adrenergic response. Hypokalemia is also a common complication that occurs from both insulin therapy and fluid repletion. Insulin administration drives extracellular potassium into cells, and fluid repletion can dilute the concentration of serum potassium. Levels of potassium should be monitored every 2 hours and repleted when potassium levels fall below 5.2 mEq/L.
Cerebral Edema
More rare but severe complications in DKA can also occur, the most worrisome of which is cerebral edema. Cerebral edema carries a high mortality rate of 21% to 90%. , Cerebral edema occurs from an elevated osmolar gradient caused by hyperglycemia, which leads to a water shift from the intracellular space to the extracellular space and cell volume contraction. Cerebral edema is more common in children and adolescents and typically presents with headache, vomiting, and decreased mental status followed by seizures, bradycardia, and respiratory arrest. Risk factors for cerebral edema include younger age, severe acidosis with low bicarbonate level on presentation, and hyponatremia with rapid hydration. Patients with cerebral edema need to be transferred to an intensive care setting and treated with intravenous mannitol.
Other Complications
Other complications in DKA management include pulmonary edema from volume repletion in patients with CKD or heart failure. Less commonly, patients may also experience rhabdomyolysis. The exact etiology of rhabdomyolysis in DKA is not known but presumed to be a combination of hyperosmolarity and hypophosphatemia. If left untreated, DKA can also lead to ischemic stroke, cerebral venous thrombosis, and hemorrhagic stroke from cerebral hypoperfusion.
PREVENTION OF DIABETIC KETOACIDOSIS
Hospitalizations for DKA have been on the rise, and preventing both the initial event as well as recurrent episodes is of utmost importance. Many patients admitted for DKA present with new- onset diabetes, most commonly in type 1 diabetes and ketosis-prone diabetes in ethnic minorities. Patients most at risk for developing type 1 diabetes include children and adolescents with a parent or sibling with type 1 diabetes and patients with other autoimmune conditions. Medical providers should be appropriately educated on these risk factors and should screen high-risk patients. If screened negative for diabetes, health care providers should continue to closely monitor their high-risk patients in addition to educating their patients and the patient’s family members on the signs and symptoms of diabetes. Research has shown that educational prevention programs on early recognition of diabetes and DKA symptoms can significantly decrease the number of children admitted with DKA at diagnosis.
Recurrent admissions for DKA must also be prevented to decrease both the morbidity and mortality associated with diabetes, as recurrent hospitalizations are associated with an increased mortality risk. Hospital readmissions are more likely to occur in patients with type 1 diabetes, but patients with type 2 diabetes can account for up to 35% of recurrent DKA cases. It is important that all patients admitted for DKA be counseled on strategies for its prevention. This includes discussing the signs and symptoms of hyperglycemia, early recognition of DKA with use of home ketone monitoring systems, and reviewing sick-day management with patients and family members. During an acute illness, patients need to establish early contact with their health care providers. Patients and family should also be counseled on the importance of continuing insulin, monitoring blood sugars, hydration, and providing correctional doses of rapid-acting insulin for hyperglycemia during acute illness.
Prevention of recurrent DKA also requires assessing the precipitating factors that may have triggered prior DKA hospitalizations. This includes assessing for insulin nonadherence related to poor health literacy, financial difficulties, substance abuse, and psychological stressors.
Insulin nonadherence may be prevented through intensive patient education and outpatient follow-up with a diabetes health care provider. Patients should also be provided with the necessary diabetes medications and supplies on discharge.
Hyperglycemic Hyperosmolar State
INTRODUCTION
The HHS is a metabolic complication of diabetes mellitus that represents a separate hyperglycemic emergency to DKA despite many similarities. HHS is characterized by severe hyperglycemia, hyperosmolarity, and dehydration in the absence of significant ketoacidosis and is more commonly found in patients with type 2 diabetes.
DEFINITION
The diagnostic criteria for HHS include hyperglycemia with a blood glucose level often exceeding 600 mg/dL, serum osmolality greater than 320 mOsm/kg, and absence of significant ketoacidosis.
PATHOPHYSIOLOGY
The pathophysiology of HHS is similar to that of DKA with some specific intricacies. At the cornerstone of both conditions is a deficiency in insulin. However, in HHS, the insulin deficiency is only relative, as the pancreas continues to make insulin but is unable to keep up with the demand needed to overcome peripheral tissue insulin resistance. In the setting of a relative insulin deficiency, the cells in the periphery enter a perceived “starvation” state leading to the release of the counterregulatory hormones glucagon, cortisol, growth hormone, and catecholamines. The counterregulatory hormones work to promote glycogenolysis and gluconeogenesis, leading to worsening hyperglycemia. The hyperglycemia then leads to a direct and indirect increase in serum osmolality. Serum osmolality is determined by the formula 2[serum sodium] + [serum glucose]/ 18 + BUN (i.e., blood urea nitrogen); therefore as the serum glucose levels rise, there will be a direct increase in the serum osmolality. Additionally, hyperglycemia triggers an osmotic diuresis, which leads to the loss of free water, glucose, and electrolytes through the urine, resulting in the indirect increase in serum osmolality concentration and worsening of dehydration.
Combined, the hyperglycemia and dehydration lead to a mixed hyponatremia. The hyperglycemia results in a hyperosmolar hyponatremia that can be corrected by decreasing the serum sodium 1.6 mEq/L per 100 mg/dL glucose rise above 100 mg/dL. In addition, the concurrent dehydration produced from osmotic diuresis leads to a hypovolemic hyponatremia that must be corrected with intravenous hydration.
A key difference in the pathophysiology of HHS in comparison to DKA is the minimal to absent production of ketones. In HHS, the pancreas continues to produce insulin, and the amount of insulin produced is sufficient to inhibit lipolysis. In the absence of lipolysis, there is no fatty acid oxidation and therefore no production of ketone bodies and resultant acidemia.
CLINICAL PRESENTATION
Clinical Findings
HHS has a clinical presentation that commonly resembles the signs and symptoms seen in DKA. Patients in HHS may present with polyuria, polydipsia, severe dehydration, weight loss, blurred vision, and changes in mental status. However, a distinguishing feature is the timing of the symptoms. Patients in DKA will often present with more acute symptoms, whereas patients in HHS may have symptoms develop over several days to weeks. A careful history should be performed to assess for any precipitating factors such as infection, cardiovascular compromise, dietary indiscretions, changes in medications, and general compliance with prescribed diabetic regimen.
On physical examination, patients may appear dehydrated with dry mucous membranes, decreased skin turgor, tachycardia, and hypotension. Unlike DKA, patients in HHS typically do not develop gastrointestinal distress. Neurologic symptoms such as decreased alteration, stupor, delirium, seizure, and focal neurologic deficits such as transient hemiplegia may also be present.
Laboratory Findings
Serum glucose will typically be above 600 mg/dL, even exceeding 1000 mg/dL in some cases. On chemistry panel, most patients will be hyponatremic with normal to elevated potassium levels. Patients will often have an elevated BUN and creatinine reflecting a prerenal azotemia. Serum osmolality will also be elevated to above 320 mOsmol/kg. In contrast to DKA, there will be an absence of significant ketoacidosis with typically normal to mildly elevated ketones, a pH greater than 7.3, and no anion gap.
TREATMENT
HHS is an endocrine emergency that requires prompt recognition and treatment. All patients must be acutely stabilized and mental status should be closely evaluated. Patients presenting with severe lethargy or stupor should be evaluated for respiratory compromise and intubated if necessary, for airway protection. Patients should be admitted to the ICU for close monitoring and care, and any precipitating causes should be investigated and treated.
Fluid Replacement
Once stabilized, patients should be treated with aggressive intravenous hydration and electrolyte replacement as needed. Isotonic fluid is preferred to restore intravascular volume depletion and decrease counterregulatory hormone action and hyperglycemia. An initial isotonic fluid bolus of 15 to 20 mL/kg during the first 1 to 2 hours of presentation, followed by 250 to 500 mL/hour, is typically recommended until glucose levels reach 300 mg/dL.
Electrolyte Replacement
Similar to DKA, patients may have depleted potassium stores from urinary losses, and potassium levels must be greater than 3.3mEq before starting an insulin drip. Potassium should also be repleted once potassium levels fall to less than 5.5 mEq/L.
Insulin
After the initial 1 to 2 hours of fluid resuscitation, an intravenous insulin drip can be started as per the preferred hospital protocol. At this time, there are no clear consensus guidelines regarding the preferred insulin drip titration protocol. Once serum glucose levels reach 300 mg/dL, isotonic fluid should be replaced by intravenous D5 ½ normal saline and continued until the patient’s mental status is restored and the serum osmolarity normalizes to less than 310 mOsmol/kg in order to maintain blood glucoses between 250 to 300 mg/dL.
During the course of HHS management, the goal is to keep glucose levels between 250 to 300 mg/dL in order to minimize the risk for cerebral edema. In animal studies, rapid correction of plasma and brain osmolality has resulted in cerebral edema. Although this has not been seen in humans, the theoretical fear of cerebral edema from osmolality overcorrection supersedes the benefit of euglycemia during HHS treatment. ,
Hypoglycemia
DEFINITION
Hypoglycemia is defined as a low blood glucose concentration below the normal range that is associated with autonomic and neuroglycopenic symptoms. Due to variable levels in which patients may exhibit symptoms of hypoglycemia, a joint American Diabetes Association and Endocrine Society work group concluded that no specific glucose threshold could be used to define hypoglycemia. In patients with diabetes, an alert value of less than 70 mg/dL was suggested to prompt attention to the patient due to the increased risk of harm from hypoglycemia by the same work group. Although any episode of hypoglycemia could be life-threatening to a patient due to its impact on cognition, severe hypoglycemia, defined as a hypoglycemic event that requires external assistance to administer carbohydrates or glucagon, can be considered an endocrine emergency.
CLINICAL PRESENTATION
Symptoms associated with hypoglycemia can be classified by their underlying cause – activation of the adrenergic system or neuroglycopenia:
- ■
Adrenergic symptoms
- ■
Heart palpitations and tachycardia
- ■
Diaphoresis
- ■
Tremor
- ■
Hunger
- ■
Nausea
- ■
Anxiety
- ■
- ■
Neuroglycopenic symptoms
- ■
Weakness
- ■
Lethargy
- ■
Altered mental status
- ■
Dizziness
- ■
Seizure
- ■
Coma/loss of consciousness
- ■
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of hypoglycemia is quite different when being considered in a patient with diabetes or without diabetes. In patients with diabetes, medications for diabetes are nearly always the cause for severe hypoglycemia. In a patient without diabetes, the differential diagnosis is broad, including accidental or intentional administration of antihyperglycemic agents, alcohol use, hepatic failure, renal failure, adrenal insufficiency, insulinoma, nesidioblastosis, bariatric surgery–related hypoglycemia, and insulin autoimmunity. For a patient with limited known history, it is appropriate to begin with a broader differential diagnosis in their initial evaluation.
EVALUATION
The first step in the evaluation of a patient is a detailed history to determine if they have the diagnosis of diabetes and, if so, a detailed history including medications. A detailed history may help in narrowing the differential diagnosis and focusing the evaluation of the patient. However, in some cases, a medical history may not be obtainable and a broader differential diagnosis and work up maybe required.
History
The history obtained for a patient with severe hypoglycemia should first focus on recent events and activities that could precipitate hypoglycemia, such as changes in food intake, physical activity, alcohol intake, and recent illnesses. A detailed list of antihyperglycemic medications, recent additions, and any dose adjustments should be reviewed. Insulin and insulin secretagogues such as sulfonylureas and meglitinides can cause a hyperinsulinemic state resulting in hypoglycemia. Other classes of medications including metformin, thiazolidinediones, SGLT-2 inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, dipeptidyl peptidase-4 (DPP-4) inhibitors, and alpha-glucosidase inhibitors do not result in a hyperinsulinemic state; however, if used in combination with insulin or insulin secretagogues, they can increase the risk of hypoglycemia. Lastly, other risk factors that can increase the risk of hypoglycemia, such as older age, longer duration of diabetes, chronic kidney disease, and malnutrition, should be assessed.
Laboratory Evaluation
In most cases of severe hypoglycemia, a capillary point-of-care glucose reading is the first available data to identify the diagnosis. Depending on the known history of the patient and the context of presentation, confirmation of hypoglycemia with measurement of blood glucose may not be necessary and should not delay treatment. However, if there is a question regarding the diagnosis of hypoglycemia, a plasma or serum glucose measurement should be obtained. Due to glycolytic enzymes in red cells and leukocytes, it is important that either sodium fluoride is added to the blood sample or the sample must be rapidly separated to prevent glycolysis and a false lowering of the measured glucose.
In the context of a limited or no history, other lab tests may be helpful in diagnosing the etiology of severe hypoglycemia including:
- ■
Insulin
- ■
Pro-insulin
- ■
C-peptide
- ■
Beta-hydroxybutyrate
- ■
Sulfonylurea and meglitinide screen
- ■
Cortisol
TREATMENT
In patients with severe hypoglycemia who require external assistance, the mainstay of treatment is the administration of glucose. The mode of administration primarily depends on the patient’s mental status and vascular access. In the inpatient hospital setting, for patients who are able to safely ingest by mouth, treatment with a rapidly absorbed carbohydrate such as glucose tablets or gel may be used. For patients who are not able to take treatment by mouth, intravenous dextrose can be administered. The typical dose is administered as an intravenous push of a 50% dextrose solution in 25-g aliquots. Patients without intravenous access can be given 1 mg of glucagon intramuscularly.
Subsequent monitoring of glucose levels following treatment is necessary after initial treatment of hypoglycemia. Glucose monitoring should immediately follow treatment of hypoglycemia, but a plan for further monitoring must also be established. The decision regarding duration and frequency for monitoring takes into account the severity of hypoglycemia, the patient’s mental status and ability to communicate, which antihyperglycemic medications were used and their duration of action, and the trend of glucose levels.
For subsequent treatment of recurrent hypoglycemia, the same treatment options may be used: oral administration of rapidly absorbed carbohydrates, intravenous dextrose, or glucagon. In some cases, a continuous intravenous infusion with a 5% or 10% dextrose may be required to maintain euglycemia. If a continuous infusion is utilized, attention must be given to the volume and respiratory status of the patient, and if receiving large volumes of fluids, electrolytes should be also be monitored.
Special Considerations
The treatment and monitoring plan for hypoglycemia depends on the medications taken by the patient. If the patient has taken acarbose, an alpha-glucosidase inhibitor, it is essential that oral treatment of hypoglycemia is with dextrose as opposed to sucrose (e.g., table sugar and candy) due to the inhibition of alpha-glucosidase in the intestine. Patients treated with insulin therapy may require prolonged monitoring and treatment depending on how recently insulin was administered and the duration of action of the insulin used. In addition, patient-specific factors such as impaired renal function and liver dysfunction, which can affect the metabolism of medications, may prolong the duration of monitoring and treatment of hypoglycemia. This especially applies to patients treated with insulin or insulin secretagogues.
PREVENTION
Prevention of hypoglycemia in patients with diabetes centers around three major themes: management of diabetes medications, patient education, and patient self-management of diabetes with blood glucose monitoring.
Following a hypoglycemic event, careful attention should be paid to evaluating the diabetes medication regimen. Medications thought to be primarily responsible for hypoglycemia should either be adjusted or removed, with appropriate alternatives prescribed. Subsequent encounters with the patient should review recurrent hypoglycemic events and the patient’s self-monitored blood glucose testing.
Patients should be educated on their diabetes medications and the potential risk for hypoglycemia. Following this education, a patient should be told which medications they should withhold or reduce doses of in the event of decreased oral intake or fasting, increased physical activity, and alcohol intake. Symptoms of hypoglycemia should be reviewed with the patient and appropriate hypoglycemia treatments prescribed, such as glucose tablets and glucagon.
Self-monitored blood glucose testing should be provided and taught to patients who are on medications with an increased risk for hypoglycemia. Patients should be instructed to test their blood glucose if they recognize signs or symptoms of hypoglycemia. However, in the event of an emergency, treatment should be administered prior to testing. In addition, depending on the medication regimen of the patient, testing their blood glucose level prior to physical activity and driving a vehicle may be recommended. For some patients, a continuous glucose monitor that is capable of alerting the patient or caregiver of hypoglycemia and impending hypoglycemia may also be effective in its prevention.
Conclusion
DKA, HHS, and severe hypoglycemia are all serious life-threatening complications of diabetes that require prompt diagnosis, treatment, and close monitoring. The diagnosis of hyperglycemic emergencies, DKA and HHS, is made based on the presence of hyperglycemia, an assessment of acid–base status, and the presence or absence of ketonemia. Treatment for both DKA and HHS include fluid resuscitation, insulin, and close monitoring of electrolytes along with treatment of any underlying precipitating factors. The diagnosis of hypoglycemia is typically made based on point-of-care glucose testing; however, it may often require laboratory confirmation and evaluation. Although the treatment of hypoglycemia is straightforward, attention must be invested in identifying the cause and any precipitating events in order to prevent its recurrence. In all diabetes emergencies, there is a strong role of patient education in the early diagnosis and prevention of each.
References
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


