Dendritic Cells
Kang Liu
Michel C. Nussenzweig
INTRODUCTION
Immunology interfaces with medicine at many points, the most prominent being infectious diseases, cancer, transplantation, allergy, and autoimmunity. Antigen-specific immunity is critical for vaccination and for protective immunity against pathogens and tumors. Moreover, when control over immune responses is lost, the result is autoimmune disease. T cells are an essential element that regulates the balance in immunity by killing infected cells, helping antibody formation, and suppressing autoimmune responses. However, T cells are incapable of recognizing native antigens. Instead, they recognize processed peptides presented by major histocompatibility complex (MHC) molecules. Dendritic cells (DCs) are professional antigen-presenting cells that inform the fight against invasive pathogens while enforcing tolerance to self- and harmless environmental antigens. They capture pathogens and receive signals from pathogens that influence the outcome of immune responses. On the basis of these signals, DCs orchestrate antigen-specific T-cell differentiation toward Th1, Th2, Th17, or Tfh pathways. Alternatively, they can silence self-reactive T cells by inducing deletion, anergy, or active regulation (via regulatory T [Treg] cells). This chapter will focus on the discovery, function, and development of DCs, and the mechanisms by which they link innate immunity to adaptive immunity.
DISCOVERY
DCs were discovered as part of an effort to understand the initiation of immunity. The mouse as an experimental animal was critical because of a system, developed by Mishell and Dutton,1 in which mouse spleen cell suspensions could be stimulated to generate antibody responses in culture. One of the early observations was that lymphocytes alone were not sufficient to induce antibody-forming cell responses and that an adherent accessory cell was required. The search for the accessory cell led to the discovery of DCs. In this section, we will review the early methods used to identify and study DCs.
Identification and Isolation
DCs were discovered by Steinman and Cohn when they examined spleen adherent cells by phase contrast microscopy.2 They then employed physical techniques to fractionate spleen cells and purify DCs. These cells were found to adhere to plastic or glass, had low buoyant density, and did not bind to erythrocytes coated with antibody. Sequential steps of density centrifugation in bovine serum albumin gradients and adherence to glass were originally used to purify DCs.3
At the time, macrophages were thought to be the key accessory cell because they composed a major population of adherent cells and also because their role in innate immunity had been long appreciated. Moreover, purified antigen loaded macrophages induced immune responses when reinjected into their hosts.4 However, macrophages failed to show robust activity in induction of antibody responses in vitro, and they rapidly degraded ingested antigens suggesting that they would be unable to present it to lymphocytes.5,6,7
Dendritic Cells Initiate T-Cell Responses
In addition to their morphology, the cell surface composition of the newly discovered DCs proved to be distinct. DCs were found to express high levels of MHC antigens.8,9 At the time, the precise function of the MHC in antigen presentation was not known, but it was already clear that the MHC was a key genetic determinant of immune responses and that it encoded many of the antigens involved in graft rejection.
The mixed leukocyte reaction (MLR) was considered an in vitro model system to study graft rejection and was used by Steinman and Cohn to examine the function of DCs.10 They found that DCs were nearly two orders of magnitude more potent than unfractionated spleen cells, B cells, or macrophages in stimulating allogeneic T cells in the MLR.10,11 On the basis of these experiments, they speculated that DCs were the accessory cells that present antigen to T cells to initiate immune responses.12 However, their conclusions were not widely accepted by immunologists because unlike traditional immune responses, the MLR did not require priming.13
Proof that DCs are antigen-presenting cells came from experiments performed by Nussenzweig and Steinman who cocultured DCs with responding T cells and hapten-modified thymocytes. They showed that DCs induced MHC-restricted cytotoxic T cells specific to the hapten and that macrophages and other purified populations of lymphoid cells were nearly inactive as accessory cells.14
Monoclonal Antibodies to Dendritic Cells
Although the morphologic and functional differences between DCs and other leukocytes were striking, few immunology laboratories were equipped to perform the purification procedures required to study DCs until monoclonal antibodies to DCs became available. A series of
mouse DC-restricted monoclonal antibodies were produced by Steinman and others starting in 1980s, including 33D1 (specific for the cell surface receptor DCIR2/Clec4a4),15 NLDC-145 (specific for the adsorptive endocytosis receptor, DEC205/cluster of differentiation [CD]205),16 and N418 (specific for the CD11c integrin).17 These monoclonal antibodies were used to establish the unique functions of DCs within heterogeneous mixtures of leukocytes and to identify DCs in situ. For example, 33D1 was used to selectively deplete DCs from mouse spleen suspensions and shows that this depleted MLR stimulating activity12 and accessory function in the Mishell-Dutton system.18
mouse DC-restricted monoclonal antibodies were produced by Steinman and others starting in 1980s, including 33D1 (specific for the cell surface receptor DCIR2/Clec4a4),15 NLDC-145 (specific for the adsorptive endocytosis receptor, DEC205/cluster of differentiation [CD]205),16 and N418 (specific for the CD11c integrin).17 These monoclonal antibodies were used to establish the unique functions of DCs within heterogeneous mixtures of leukocytes and to identify DCs in situ. For example, 33D1 was used to selectively deplete DCs from mouse spleen suspensions and shows that this depleted MLR stimulating activity12 and accessory function in the Mishell-Dutton system.18
Dendritic Cell Subsets
Shortman and colleagues first showed that DCs are heterogeneous and suggested the existence of DC subsets.19,20 These subsets differ in their anatomic distribution, cell surface marker expression, and function. In the mouse, three major groups of DCs exist in the steady state: plasmacytoid DCs (pDCs), conventional DCs (cDCs), and migratory DCs (mDCs). pDCs are important mediators of antiviral immunity through their ability to produce large amounts of type I interferons (IFNs) upon viral infection (see following discussion). cDCs are composed of two major subsets, namely CD8α+ and CD8α-.21 mDCs are present in nonlymphoid tissues such as the liver, gut, skin, lung, and aorta, and they are composed of two main subsets CD103+ and CD103-.22 These nonlymphoid tissue DCs are referred to as mDCs because they transit from tissue to lymphoid organs.21,23
The two major subsets of mouse spleen cDCs have some overlapping functions. Both can process and present antigens to T cells and also secrete cytokines such as interleukin (IL)-12, which can influence the ultimate polarization of the T-cell response to pathogens. However, the two subsets also have unique functions in vivo and are not redundant.24,25,26
Some of the differences in cDC subset function may be accounted for differential expression of toll-like receptors (TLRs) or other mediators of cDC activation. For example, spleen CD8α+ cDCs express TLR3 (recognizing doublestranded ribonucleic acid [RNA]) but lack TLR5 (recognizing flagellin) and TLR7 (recognizing single-stranded RNA), whereas CD8α- cDCs express TLR5 and TLR7 but have low levels of TLR3.27 Comparative analysis of messenger RNA expression profiles between the two DC subsets revealed that the two are as different from each other as are T- and B-lymphocytes.28 This is in part reflected in the antigen processing capacity of the two cDCs types. CD8α+ cDCs are specialized for MHCI cross-presentation, and enriched in Tap1, Tap2, calreticulin, calnexin, Sec61, ERp57, ERAAP, as well as cystatin B and C, all of which are involved in MHCI presentation or inhibition of enzymes that process peptides for MHCII presentation. In contrast, CD8α- cDCs, which are biased for MHCII presentation, were enriched in cathepsins C, H, and Z, asparagine endopeptidase, GILT, and H2-Mbeta 1, all of which are implicated in the MHCII antigen-processing pathway.28 The immunologic consequences of this specialization can be seen after immunization with antigens targeted to one or the other cDC in vivo. Antigens targeted to CD8α+ and CD8α- DCs by chimeric monoclonal antibodies that bind endocytic receptors Dec205/CD205 or DCIR2/Clec4a4 are biased to strong CD8 or CD4 T-cell responses, respectively.28
In conclusion, the two major subsets of DCs have distinct microanatomic locations, gene expression profiles, and functions. However the CD8α+ cDC in lymphoid tissue and CD103+ subset in nonlymphoid tissue DCs subsets are far better characterized than the CD8α- cDC or CD103- subset, which are heterogeneous and difficult to distinguish from activated monocytes.
ANATOMIC DISTRIBUTION
Lymphoid Organs
Peripheral lymphoid organs (ie, spleen, lymph nodes, and mucosal associated lymphoid tissues) are the sites where primary immune responses develop, including activation of helper, killer, and antibody-forming cells. Initiation of immune responses is facilitated by the highly organized structure of the peripheral lymphoid organs, with lymphocytes segregated into B- and T-cell areas. In addition to lymphocytes, the T-cell area also contains large interdigitating cells that were initially thought to be macrophages. However, cytologic and functional studies of spleen and lymph nodes in rat and mice revealed that interdigitating cells lack phagosomes and lysosomes, and are poorly phagocytic.29,30,31 These features distinguished interdigitating cells from macrophages and suggested that they might be related to the cDCs isolated from mouse spleen.
Mouse interdigitating cells were later proven to be cDCs based on immunohistochemistry using DC-specific monoclonal antibodies. In human lymphoid organs, interdigitating cells in the T-cell area express CD83, a member of immunoglobulin superfamily. This marker is not conserved in the mouse where the interdigitating cells in the T-cell area of lymph nodes express CD11c and DEC205/CD205. In contrast, the two cDC subsets in the mouse spleen segregate into distinct microanatomic compartments. CD8α+ cDCs in spleen T-cell areas resemble those in the lymph nodes in that they express DEC-205, whereas those in the bridging channels and red pulp express DCIR2.28
In the T-cell area, cDCs form a dense network of cells that are primarily sessile but constantly protrude and retract large membrane folds into their environment.32 T cells actively migrate through the DC network in search of antigen, and when they find a cDC presenting cognate antigen, they pause for a prolonged interaction.33 The large DC membrane protrusions are thought to increase the surface area for antigen presentation and facilitate antigen detection by migrating T cells.34
Nonlymphoid Organs
In addition to the lymphoid organs, DCs are also found in most nonlymphoid organs and in all epithelial surfaces that contact the environment. In organs such as heart, lung, kidney, the dermal layer of skin, and meninges and choroid
plexus in the brain, they are found in interstitial spaces that are drained by lymphatics.35,36,37 Like their lymphoid counterparts, interstitial DCs can be distinguished from macrophages by abundant expression of MHCII and low levels of lysosomal hydrolases. In comparison to macrophages, which are also abundant in the interstitial spaces, interstitial DCs are less phagocytic and more sensitive to radiation. Tissue macrophages and DCs can be distinguished by immunohistology; the former typically express F4/80 and lysosomal hydrolase, whereas the latter express MHCII and CD11c. The standard markers for distinguishing DCs and macrophages in interstitial tissues are listed in Table 16.1. Importantly, although these markers help to differentiate macrophage and cDCs in most tissues, they are not sufficient in defining cell lineages unless used in combination with functional and developmental analysis. This is because CD11c and MHCII can also be expressed by lung macrophages and induced on monocytes during inflammation.38
plexus in the brain, they are found in interstitial spaces that are drained by lymphatics.35,36,37 Like their lymphoid counterparts, interstitial DCs can be distinguished from macrophages by abundant expression of MHCII and low levels of lysosomal hydrolases. In comparison to macrophages, which are also abundant in the interstitial spaces, interstitial DCs are less phagocytic and more sensitive to radiation. Tissue macrophages and DCs can be distinguished by immunohistology; the former typically express F4/80 and lysosomal hydrolase, whereas the latter express MHCII and CD11c. The standard markers for distinguishing DCs and macrophages in interstitial tissues are listed in Table 16.1. Importantly, although these markers help to differentiate macrophage and cDCs in most tissues, they are not sufficient in defining cell lineages unless used in combination with functional and developmental analysis. This is because CD11c and MHCII can also be expressed by lung macrophages and induced on monocytes during inflammation.38
TABLE 16.1 Cell Surface Comparison between Dendritic Cells and Macrophage | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Afferent Lymphatics
Afferent lymphatics in rats, rabbits, guinea pigs, sheep, and humans contain cells with motile processes that were named veiled cells because of their large membrane protrusions.34,39,40,41 These cells were found to be poorly phagocytic, express high levels of MHCII, and are potent stimulators of the MLR in vitro. Lymphadenectomy and thoracic duct cannulation in larger animals such as rats and sheep made it possible to collect these cells from afferent lymph. Using this technique, Huang et al. revealed that veiled cells are constantly carrying intestinal epithelial contents via lymphatics to the cDC network in the T-cell area of the mesenteric lymph nodes.42 Therefore, in the healthy individual, these veiled cells continually carry and present self- and harmless antigen to the adaptive immune system, thereby contributing to the induction of peripheral tolerance (see following discussion); during infection, they carry pathogen-derived antigens and induce specific T-cell activation. Thus veiled cells correspond to mDCs.
Dendritic Cells in the Skin and Other Body Surfaces
Several types of DCs, monocytes, and macrophages exist in the skin and at other body surfaces. Professional antigenpresenting cells in the skin include Langerhans cells (LCs) in the epidermis, and CD103+ and CD103- DCs in the dermis.
DENDRITIC CELL DEVELOPMENT
Origin of Conventional Dendritic Cells in the Lymphoid Organs
DCs, like all other leukocytes, develop from bone-marrow-derived hematopoietic stem cells. Monocytes, macrophages, granulocytes, megakaryocytes, and erythrocytes differentiate from a common myeloid progenitor (CMP) whereas B, T, and natural killer (NK) cells differentiate from a common lymphoid progenitor.43,44,45 Although early cell transfer and genetic experiments were interpreted to indicate that DCs originated from lymphoid and myeloid progenitors,46,47,48,49 subsequent work showed that all DCs are derived from myeloid progenitors.50
Relationship between Dendritic Cells and Monocytes
In the steady state, DCs and monocytes can readily be distinguished based on cell surface marker expression. For example, DCs are CD11c+CD115-/loMHCII+ and monocytes are CD11b+CD115+F4/80+ (see Table 16.1). In contrast, under inflammatory conditions or in tissues such as lung and intestine, where they can be exposed to pathogenic and nonpathogenic microbes, monocytes develop many of the characteristic features associated with DCs, and the distinction becomes far more difficult. Moreover, the question of how DCs are related to monocytes and macrophages during development was a vexing problem that was only resolved very recently.
The idea that monocytes might be the direct precursors of DCs in vivo was suggested by elegant experiments with human CD14+ monocytes.51 Monocytes undergoing reverse transmigration, which simulates their entry into lymphatic vessels from tissues, lose expression of monocyte markers CD14 and CD64 and upregulate human leukocyte antigen-DR and CD54.51 Phagocytosis further stimulates expression of CD80, CD86, human leukocyte antigen-DR, DC-LAMP, and CD83, all of which are expressed by DCs.51 Consistent with these cell surface changes, activated monocytes stimulate allogeneic T-cell proliferation in MLRs.
Similarly, in mice, microspheres injected intracutaneously are phagocytosed by CD11b+ F4/80+ monocytes that can then be found in the draining lymph nodes 3 to 4 days after the injection.52 These cells display high expression of MHCII, MHCI, CD86, and stimulate allogeneic T-cell proliferation; however, unlike DCs, they express low levels of CD11c and no CD8α. Therefore, monocytes in the skin did not appear to become classical cDCs upon migration to lymph nodes; nevertheless, these activated monocytes shared many of the features associated with DCs. Finally, activated monocytes that appear in the spleen during Listeria monocytogenes infection expressed many of the cell surface features of cDCs.53 These cells produce tumor necrosis factor (TNF) and inducible nitric oxide synthase (iNOS) and were named TNF/iNOS-producing DCs (Tip-DCs).53 Tip-DC deficiency led to lethal L. monocytogenes infection; however, this effect was independent of the development of adaptive CD8 T-cell immunity.53
The idea that DCs develop from monocytes was tested by several laboratories in direct transfer experiments in
mice without clear success.54,55,56,57 Indeed, it was always the nonmonocyte fraction of the bone marrow that produced cDCs.54,56 The only exception to this rule occurred after inflammation induced by Freund’s Complete Adjuvant (CFA), when monocytes differentiated into CD11c+CD11b+Mac3+ cells, which again are phenotypically distinct from cDCs.56
mice without clear success.54,55,56,57 Indeed, it was always the nonmonocyte fraction of the bone marrow that produced cDCs.54,56 The only exception to this rule occurred after inflammation induced by Freund’s Complete Adjuvant (CFA), when monocytes differentiated into CD11c+CD11b+Mac3+ cells, which again are phenotypically distinct from cDCs.56
The inability of monocytes to produce cDCs in steadystate lymphoid organs was further confirmed in a series of elegant genetic studies using reporter mice carrying Rosa26-StopfloxEGFP.58 In these mice, a lox-flanked Stop transcription signal must be deleted by Cre expression before GFP can be transcribed. Breeding LysM-Cre and Rosa26-StopfloxEGFP mice results in lysozyme promoter-driven Cre expression in monocytes and neutrophils, as well as deletion of the Stop sequence from Rosa26-StopfloxEGFP in these cells. As a result, monocytes and their progeny are irreversibly marked with enhanced green fluorescent protein expression. Classical DCs in the peripheral lymphoid organs remained enhanced green fluorescent protein negative in these mice; therefore, they cannot be derived from monocytes.58
In conclusion, experiments in humans and mice show that monocytes can develop some of the features of DCs under conditions of inflammation in vivo, or when cultured with cytokines in vitro, but they are not precursors of cDCs.
Dendritic Cell Progenitors in the Bone Marrow
If monocytes do not produce DCs, the question remains: what is the origin of DCs and where does commitment to this lineage occur? The first clue in solving this problem came from the identification of a common precursor for monocytes, macrophagesm and classical DCs (macrophage-DC progenitors [MDPs]54). MDP are Lin-CX3CR1+CD11b-CD 115+cKit+CD135+ and account for 0.5% of all bone marrow mononuclear cells in mice.54,59 When cultured with granulocyte-macrophage-colony stimulating factor (GM-CSF) in vitro or adoptively transferred into mice, these cells produced macrophages and DCs, but not neutrophil granulocytes, B-or T-lymphocytes, or NK cells.54,59,60 Therefore, MDPs are more restricted than CMPs, from which they are derived.
A DC-restricted progenitor that produces cDCs and pDCs but not monocytes in vitro61 or in vivo62 was then identified based on reduced cKit (CD117) and residual Flt3 expression and named common-DC progenitor (CDP; Lin-CD115+Flt3+CD117lo). The CDP is downstream of CMP and MDP because adoptive transfer of either CMP or MDP gives rise to CDP and monocytes.60 More importantly, these experiments showed that the split between the monocyte and DC lineages occurs in the bone marrow between the MDP and CDP stages of development.60
Migratory Pre-Dendritic Cells
cDC precursors must migrate from the bone marrow to the lymphoid organs through the blood. However, MDPs and CDPs are restricted to the bone marrow.60 The identity of the DC precursor was suggested by the finding that lowdensity CD11c+MHC II-SIRPαlo cells isolated from blood, bone marrow, and periphery had the potential to produce cDCs.56,63,64 Although this group of cells was heterogeneous, the combination of these markers and persistent expression of Flt3 defined pre-DCs and allowed for their isolation in mice.60
Pre-DCs migrate from the bone marrow to the blood and then to peripheral lymphoid organs and nonlymphoid tissues.60 These cells comprise ˜0.5% of all leukocytes in bone marrow, 0.02% in blood, 0.05% in the spleen, and 0.03% in the lymph nodes. Pre-DCs have a short half-life in the blood of < 1 hour65; this, together with the small number of these cells in blood and tissues, may explain why previous efforts to identify pre-DCs failed and why human pre-DCs have yet to be identified.
Pre-cDCs isolated from the bone marrow, blood, or spleen give rise to both CD8α+ and CD8α- cDCs in lymphoid and nonlymphoid organs.60 Thus, the pre-DC is a progenitor with significant plasticity. In conclusion, the DC and monocyte lineages split in the bone marrow, where MDPs give rise to both monocytes and CDP; the latter produce pre-DCs, which migrate from bone marrow through the blood to the periphery to give rise to DCs (Fig. 16.1).
Origin of Nonlymphoid Tissue Dendritic Cells
Skin
The skin contains LCs in the epidermis and CD103+ and CD103- DCs in the dermis.
Epidermis. LCs were discovered in 1868 by Paul Langerhans. Although LCs were initially thought to be of neural crest origin,66,67 this view was changed with the discovery that LCs are closely related to leukocytes,68,69 express Fc receptors70 and MHCII antigens,71 and are competent to present antigen to primed T cells.72
Experiments using bone marrow chimeras and parabiotic mice demonstrated that LCs are long-lived cells that divide in situ in the skin.73 These cells originate from fetal liver-derived progenitors in an Flt3L-independent manner, and they are only replaced by bone marrow-derived hematopoietic cells during inflammation.73,74 Thus, the origin of the LCs is different from that of cDCs.
Nevertheless, mouse LCs have been valuable as models for studying the differentiation and migration of antigenpresenting cells.75,76,77,78,79,80,81 LCs in the epithelial layer of the skin can take up particles82 and contain distinct granules, known as Birbeck granules, which are a type of endocytic organelle enriched in langerin.83,84 While LCs express Fc receptors and MHCII in situ, the expression of Fc receptors drops and MHCII molecules redistribute from intracellular compartments to the cell surface in cultured LCs that become more highly immunostimulatory.75,76,85 Cells that resemble LCs are also found in other stratified squamous epithelia such as the vagina, cervix, anus, pharynx, and esophagus.
LCs show many of the phenotypic features of cDCs, including morphology, ability to redistribute large amounts of MHCII from the endocytic system to the cell surface, and capacity to stimulate allogeneic T-cell proliferation in vitro after activation.75,78,86,87,88 Importantly, however, LCs differ from DCs in that they are resistant to irradiation, they are self-renewing in situ and not of pre-DC origin in the steady
state, and finally, like monocytes and macrophages, their development is dependent on macrophage-colony stimulating factor (M-CSF) and not on Flt3L.89 LC precursors, which are CX3CR1+CD45+, colonize the dermis during late embryonic development.90,91 In mice, these precursors complete their differentiation into LCs by postnatal d2, whereupon they undergo a burst of cell division between postnatal d2 and d7, which accounts for the dramatic increase in LCs during this developmental window. The LC precursor in skin appears to resemble the MDP.90 Recently, fate-mapping analysis revealed that LCs derive from primitive macrophages during yolk sac stage of embryogenesis, and not from postnatal hematopoietic progenitors.91 These two lineages can be differentiated by their dependence of Myb gene. The absence of Myb impaired HSC development, but did not affect development of the yolk sac macrophage progenitor that gives rise to LCs, microglia and liver Kupffer cells. This finding places the LC in its own differentiation pathway distinctive from cDC and monocyte.91
state, and finally, like monocytes and macrophages, their development is dependent on macrophage-colony stimulating factor (M-CSF) and not on Flt3L.89 LC precursors, which are CX3CR1+CD45+, colonize the dermis during late embryonic development.90,91 In mice, these precursors complete their differentiation into LCs by postnatal d2, whereupon they undergo a burst of cell division between postnatal d2 and d7, which accounts for the dramatic increase in LCs during this developmental window. The LC precursor in skin appears to resemble the MDP.90 Recently, fate-mapping analysis revealed that LCs derive from primitive macrophages during yolk sac stage of embryogenesis, and not from postnatal hematopoietic progenitors.91 These two lineages can be differentiated by their dependence of Myb gene. The absence of Myb impaired HSC development, but did not affect development of the yolk sac macrophage progenitor that gives rise to LCs, microglia and liver Kupffer cells. This finding places the LC in its own differentiation pathway distinctive from cDC and monocyte.91
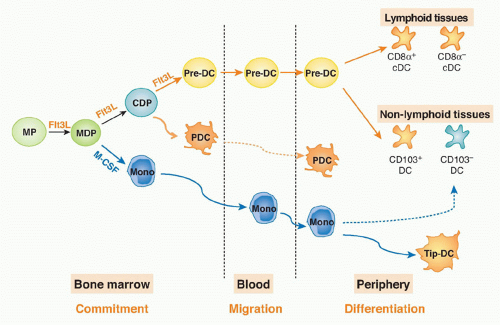 FIG. 16.1. Dendritic Cell and Monocyte Origin and Development. |
Dermis. The dermal layer of the skin contains two subsets of DCs: CD103+ and CD103- DCs. CD103+ dermal DCs are langerin+CD11blo, whereas CD103- dermal DCs are langerin-CD11b+.92,93,94,95 Although both LCs and CD103+ dermal DCs express langerin, CD103+ dermal DCs are more closely related to spleen CD8α+ cDCs than to LCs.25,37,96 CD103+ dermal DCs, like spleen cDCs, are M-CSF-independent and Flt3-dependent.37 Unlike LCs that can self-renew in situ, dermal CD103+ DCs are continually replenished from the same bloodborne pre-DCs that give rise to lymphoid organ DCs.95 In addition, like CD8α+ splenic cDCs, development of CD103+ dermal DCs requires expression of the IFN regulatory factor (IRF)8, Id2, and Batf3 transcription factors (see following discussion). Finally, both dermal CD103+ dermal DCs and CD8α+ cDCs specialize in antigen cross-presentation.28,96 The CD103- dermal DC compartment appears to be a heterogenous mixture of cells, some of which are Flt3 dependent and of pre-DC origin and others M-CSF-dependent and of monocyte origin.37
Skin-draining lymph nodes contain subpopulations of DCs that resemble CD103+ and CD103- dermal DCs.92,95 Their migration from the skin to the draining lymph nodes is dependent on CCR7 but independent of CD62L. In the steady state, these cells account for 20% of lymph node DCs while the remainder of the DCs are derived from blood pre-DCs.60 Inflammation dramatically enhances the migration of dermal DCs to the lymph node.52,58
Gut, Kidney, Lung, and Liver
Similar to the skin, CD103+ and CD103- DCs are found in nearly all mouse nonlymphoid tissues.37,97 However, the origin of these cells was not established until very recently. CD103+ nonlymphoid tissue DCs express Flt3, a marker expressed on most cells in the DC lineage but not on monocytes. Furthermore, mice deficient in Flt3L or its receptor, Flt3, showed significant decreases in CD103+ nonlymphoid tissue DCs but normal numbers of monocytes.37,98 Finally, adoptive transfer of pre-DCs into naïve recipients, or mice in which CD11c cells had been depleted, resulted in reconstitution of the CD103+ nonlymphoid tissue DC compartment in the liver, lung, kidney, and intestine.37,57 In contrast, adoptive transfer of 50 times more monocytes failed to show detectable DC progeny in naïve mice.37,57 Thus, in the steady state, CD103+ DCs in all tissues tested are pre-DC
and not monocyte-derived. Only after depletion of endogenous CD11c+ cells were monocytes able to produce some CD103- DCs.37,57 The idea that all CD103+ nonlymphoid tissue DCs are derived from pre-DCs and not from monocytes is further supported by genetic experiments that show a common requirement for Batf3, Id2, and IRF8 for lymphoid tissue CD8α+ DCs, and CD103+ nonlymphoid tissue DCs, but not monocytes or macrophages (see following discussion).24,37,99,100
and not monocyte-derived. Only after depletion of endogenous CD11c+ cells were monocytes able to produce some CD103- DCs.37,57 The idea that all CD103+ nonlymphoid tissue DCs are derived from pre-DCs and not from monocytes is further supported by genetic experiments that show a common requirement for Batf3, Id2, and IRF8 for lymphoid tissue CD8α+ DCs, and CD103+ nonlymphoid tissue DCs, but not monocytes or macrophages (see following discussion).24,37,99,100
In conclusion, in addition to DCs in the lymphoid organs, pre-cDCs also contribute to CD103+ DC development in nonlymphoid tissues. CD103- DCs appear to be more heterogenous; some of these cells are derived from pre-DCs while others may be of monocyte origin.
Plasmacytoid Dendritic Cells
pDCs were described by pathologists as a unique population of cells in the peripheral lymphoid organs that resemble plasma cells and monocytes and were referred to as plasmacytoid monocytes. In 1990s, Svensson et al. described a population of cells in human blood that produces large amounts of type I IFN upon exposure to herpes simplex virus, and they named these cells IFN-producing cells.101 Both of these populations correspond to the pDCs described by Liu and colleagues.102,103,104
pDCs were first purified from human tonsil and blood on the basis of being CD4+CD3-CD11c-. They were then shown to develop DC-like morphology upon culture with IL-3 and anti-CD40. Liu and colleagues discovered that purified pDCs produced 100 to 1,000 times more type I IFN than the other blood cell types following viral activation.104 Subsequently, pDCs were identified and isolated from lymphoid tissues in mice, and later in rats, monkeys, and pigs. In humans, the pDCs express CD4, MHCII, CD68, CD123, and blood DC antigen 2 (BDCA2), but do not express other lineage marker such as CD3 (T cell), CD14 (monocyte), CD19 (B cell), CD16 and CD56 (NK cell), or CD11c, BDCA1, and BDCA3 (cDC). They are identified as CD4+, CD11c-, and Lin- (CD3, CD14, CD16, CD19, CD56) cells.105 In mice, pDCs express CD11c (lower levels than cDCs), Gr1, B220, and PDCA1; low levels of MHCII; do not express CD11b or CD19;and they are frequently isolated by virtue of CD11c and B220 expression.106
pDCs are closely related to cDCs; they originate from the same progenitor, the CDP. Development of pDCs is also critically dependent on Flt3L. Unlike cDCs, however, pDCs do not express high levels of MHC II in the steady state and therefore, their primary function is not antigen presentation to CD4 T cells. Instead, pDCs focus on sensing infection and producing type I IFN. Depletion of pDC blocks antiviral IFN responses but also results in loss of Treg homeostasis, impaired differentiation of Th cells, and initiation of CD8 T-cell responses (see following discussion).107,108
Growth Factors for Dendritic Cells and Monocytes
GM-CSF, M-CSF and Flt3L are essential growth factors for myeloid cell development in vitro and in vivo. During the early stages in myeloid development, shared progenitors of lymphocytes, polymorphonuclear leukocytes, monocytes, and DCs express one or more of the receptors for these growth factors. For example, Flt3 is expressed very early in hematopoiesis on shared progenitors of lymphocytes and all myeloid cells.109 However, with the exception of pre-DCs, Flt3 expression is lost by nearly all leukocytes by the time they enter circulation.60 Conversely, M-CSF receptor expression is retained by cells in the monocyte lineage but lost on pre-DCs and DCs.60
As might be predicted from their patterns of expression, M-CSF and Flt3L are essential for normal development of the monocyte and DC lineages, respectively. For example, mice deficient in M-CSF or its receptor show deficiencies in monocyte, macrophage, and LC development but have normal spleen and lymph node DCs.110 In contrast, DC development is impaired in mice deficient in either Flt3L or Flt3, but monocyte development is normal.59,111 Consistent with these observations, administration or overexpression of Flt3L results in selective expansion of DC populations, including fully differentiated DCs in lymphoid organs.59,112
Dendritic Cell Cultures
In the early 1990s, Banchereau and Schuler and their colleagues established in vitro culture systems to produce human monocyte-derived DCs (moDCs), and Inaba and Steinman for the mouse equivalent.76,113,114 These culture systems are widely used today for basic and clinical studies. Bone marrow cells from mice or monocytes from humans are cultured for 6 days in medium containing GM-CSF to produce cells with dendritic morphology that exhibit modest phagocytic activity, express CD11c and MHCII, and stimulate the MLR. Differentiation to moDCs is further stimulated by addition of lipopolysaccharide, which induces a series of changes that were originally described as maturation. Notably, monocytes do not give rise to DCs in the lymphoid organs in vivo (see previous discussion), and GM-CSF is entirely dispensable for DC development in vivo115; thus, the moDCs generated in GM-CSF culture are not entirely representative of the cDCs found in the lymphoid tissues. moDCs may be more closely related to activated monocytes than to steady-state DCs in the lymphoid organs.
Authentic cDCs can be produced in in vitro cultures of bone marrow cells supplemented with Flt3L, which is an essential growth factor for DCs in vivo.116 Flt3L cultures produce all of the known subsets of DCs, including cells with features of pDCs, CD8α+/CD103+ cDCs, and CD8α- cDCs.116
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


