Dementia Including Alzheimer’s Disease: Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder projected to afflict more than 13 million Americans and 81 million individuals worldwide in the next 40 to 50 years. The disease is characterized by progressive cognitive and behavioral deficits accompanied by diffuse structural abnormalities in the brain. AD is associated with significant morbidity and mortality and is currently among the 10 most common causes of death in the United States. Unfortunately, the devastations of AD affect both the patient and caregivers, and involve numerous aspects of human life including physical, socioeconomic, psychological, functional, and quality of life. In addition, caring for patients with AD places heavy financial burden on patients, families, and the health care system at large. In the United States, the average annual cost of managing a patient with AD varies between $18,400 and $36,000, while, as a nation, we spend close to $100 billion each year to care for patients with AD. Similar data are reported from other countries as well. In the United Kingdom, more than $8 billion or 0.6% of the gross domestic product is spent each year to care for patients with cognitive impairments. Recognizing the enormity of the burden of AD, international collaborations between clinicians, researchers, policy makers, patient advocacy groups, the media, and many others have increased public awareness of the global impact of the disease and have laid the foundation for the development of effective preventive and therapeutic strategies as well as improvements in care management for patients with AD.
Recent advances in biomedical research have provided critical knowledge related to the pathobiology of AD and mechanisms underlying neuronal plasticity and death. While current therapies work mainly to slow the inevitable progression of the disease, these newly identified mechanisms now serve as targets to develop effective novel therapies that could either delay or preferably arrest the progression of AD. Moreover, treatment strategies are being developed for primary prevention of AD, especially for individuals at increased risk for the disease. The present chapter includes information on the major aspects of AD, including the pathology, epidemiology, clinical presentations, diagnosis, management, and potential neuroprotective therapies. Details related to non-Alzheimer types of dementia are discussed in Chapter 67.
Genetics of Alzheimer’s Disease
Alzheimer’s disease is characterized by progressive cognitive and behavioral deficits accompanied by diffuse structural abnormalities in the brain. The common symptoms of the disease include memory loss, confusion, impaired judgment, personality changes, disorientation, and loss of language skills. Based on the onset of symptoms, AD is normally divided into two groups: early- (<60 years) and late-onset (>60 years). Early-onset, also called familial AD (FAD), accounts for approximately 3% of all the cases and has to date been linked to mutations in the genes for the amyloid precursor protein (APP; gene name APP) on chromosome 21, presenilin 1 (PS1; gene name PSEN1) on chromosome 14, and presenilin 2 (PS2; gene name PSEN2) on chromosome 1. Among these genes, more than 160 different mutations have so far been identified, accounting for approximately 40% of all cases of FAD. Most of the mutations (∼140) are found in the PSEN1 gene, while the remaining are almost equally split between APP and PSEN2. FAD is characterized by early onset (from mid 30s to mid 50s) of cognitive symptoms, but is clinically indistinguishable from late-onset AD.
Late-onset AD (LOAD), also called sporadic AD, accounts for close to 97% of all cases of AD and to date has not been linked with any mutations. Aging is the single most important risk factor for LOAD. Additionally, LOAD has been associated with many other environmental and genetic risk factors, including hypercholesterolemia and other vascular risk factors, history of head trauma, and low educational attainment. Among the genetic risk factors, the ϵ4 allele of the apolipoprotein E (APOE) gene on chromosome 19 is the only polymorphism consistently found associated with LOAD in both case–control and genetic studies. The APOE gene exists as three major alleles (ϵ2, ϵ3, and ϵ4) that encode three different ApoE isoforms: ApoE2, ApoE3, and ApoE4. Interestingly, these isoforms only differ in amino acid sequences either at position 112 and/or 158: E2 (cysteine 112, cysteine 158), E3 (cysteine 112, arginine 158), and E4 (arginine 112, arginine 158). The inheritance of the ϵ4 allele confers a major risk for developing AD, while the ϵ2 allele confers protection. For example, presence of one copy of the ϵ4 allele increases risk of AD by fourfold, whereas inheritance of two copies enhances the risk by almost ninefold. However, unlike genetic mutations associated with FAD, the presence of APOE4 (or other polymorphisms) is neither necessary nor sufficient to cause AD. Even though the first report of an association between APOE4 and AD was published over a decade ago, the precise molecular mechanism(s) underlying this association still remain elusive. It is currently unknown if the APOE4 allele influences the rate of production, clearance, or aggregation of amyloid β-peptide or whether it influences cholesterol metabolism and inflammation that reportedly play a major role in the pathobiology of AD.
Neuropathology of Alzheimer’s Disease
The neuropathological hallmarks of AD include amyloid plaques, neurofibrillary tangles, and amyloid angiopathy accompanied by diffuse loss of neurons and synapses in the neocortex, hippocampus, and other subcortical regions of the brain (Figure 65-1). The predominance of the amyloid plaques versus neurofibrillary tangles or amyloid angiopathy can differ from one patient to another. However, the neuronal/synaptic loss is a constant feature and eventually the direct cause of dementia. Interestingly, the distribution of the disease pathology seems to follow a region-specific pattern with the amyloid plaques more prevalent in the neocortex and the neuronal/synaptic loss more prevalent in the hippocampus, posterior cingulate, and corpus callosum areas of the brain closely involved with memory formation and higher cortical activities. The amyloid angiopathy is often observed in the “classical” LOAD, but seems to be more prevalent in a small subgroup of FAD (Dutch E693Q and Iowa D694N mutations in the APP gene). Finally, AD brains are also characterized by a diffuse and widespread invasion of reactive astrocytes, mostly concentrated in the hippocampus and around areas of neuronal loss; however, this is not specific to AD and can be observed in other neurodegenerative disorders associated with inflammatory and neurotoxic insults.
Figure 65-1.
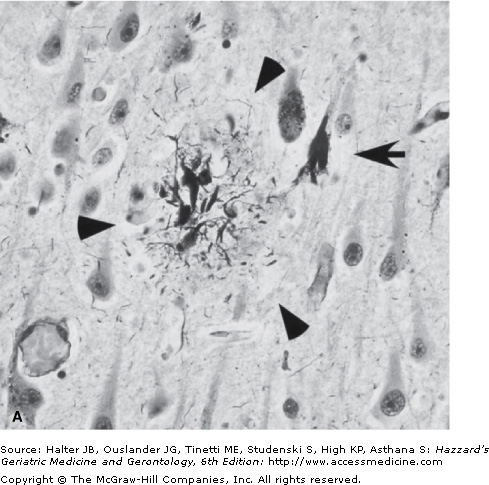
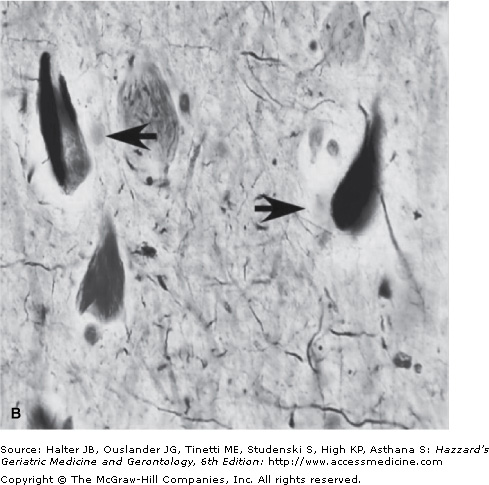
Small section of the neocortex from a patient with Alzheimer’s disease showing two classical neuropathological lesions of the disease. (A) The modified silver staining shows one dense senile (amyloid) plaque indicated by three arrowheads. The plaque consists of aggregated extracellular deposits of Aβ fragments surrounded by silver-positive dystrophic neuritis. The arrow indicates a neuron containing neurofibrillary tangles, which appear as dark masses of abnormal filaments occupying most of the cytoplasm. (B) The image shows higher magnification of two neurons containing neurofibrillary tangles (indicated by arrows). (Photograph courtesy of Shahriar Salamat, MD, PhD, University of Wisconsin School of Medicine and Public Health, Department of Pathology and Laboratory Medicine.)
The dominant component of the plaque core is amyloid β-peptide (Aβ) organized in fibrils of approximately 7 to 10 nm intermixed with nonfibrillar forms of the peptide (Figure 65-1). The most characteristic form of the amyloid plaque, the “neuritic plaque”, is characterized by a dense core of aggregated fibrillar Aβ, surrounded by dystrophic dendrites and axons, activated microglia, and reactive astrocytes. In addition, diffuse deposits of Aβ, likely representing a prefibrillary form of the aggregated peptide, are found without any surrounding dystrophic neurites, astrocytes, or microglia. These diffuse plaques can be found in limbic and association cortices, as well as in the cerebellum.
The other neuropathological hallmark of AD is a neurofibrillary tangle (NFT) found exclusively in the cytoplasm of neurons (Figure 65-1). The tangles appear as paired, helically twisted protein filaments composed of highly stable polymers of cytoplasmic proteins called tau. Tau comprises a group of alternatively spliced proteins found in the cytoplasm that possess either three or four microtubule-binding domains and can assemble with tubulin, thus helping the formation of cross bridges between adjacent microtubules. Tau proteins can be phosphorylated in multiple sites and the degree of phosphorylation is inversely correlated with binding to microtubules. As a result, highly phosphorylated tau proteins dissociate from microtubules and polymerize into filaments forming NFTs. In addition to AD, the abnormal accumulation of filamentous tau is observed in frontotemporal forms of dementia, progressive supranuclear palsy, corticobasal degeneration, and Pick disease. Contrary to prior belief, tau proteins themselves can cause dementia, and multiple mutations in the tau gene have been found in frontotemporal dementia with parkinsonism. The possible role of tau proteins in the pathogenesis of AD and their potential interaction with Aβ are still unclear and are discussed in the next section.
Pathobiology of Alzheimer’s Disease
Aβ is a 39 to 43 amino acid hydrophobic peptide proteolytically released from a much larger precursor, the amyloid precursor protein (APP). The generation of Aβ from APP (Figure 65-2) requires the sequential recruitment of two enzymatic activities: β-secretase, also called BACE1 (beta-site APP cleaving enzyme 1), and γ-secretase, a multimeric protein complex containing presenilin, nicastrin, Aph-1, Pen-2, and CD147. The β cleavage is the rate-limiting step and occurs before the γ cleavage. It liberates a large N-terminal fragment of the protein (sβAPP) that is released in the extracellular milieu and a small (∼12 kDa) membrane-anchored fragment called β-APP-CTF (or C99). The release of the large N-terminal domain allows subsequent γ cleavage, and liberation of Aβ and the signaling of active intracellular domain of APP (Figure 65-2). Generation of Aβ40 and Aβ42 results from γ cleavage of Aβ at positions 40 and 42, respectively. The release of Aβ in the extracellular milieu is followed by oligomerization and aggregation in the form of fibrils and amyloid plaques. Additionally, small Aβ aggregates are also found in the soma of the neurons suggesting that the Aβ fragments can escape secretion and aggregate in the intracellular environment.
Figure 65-2.
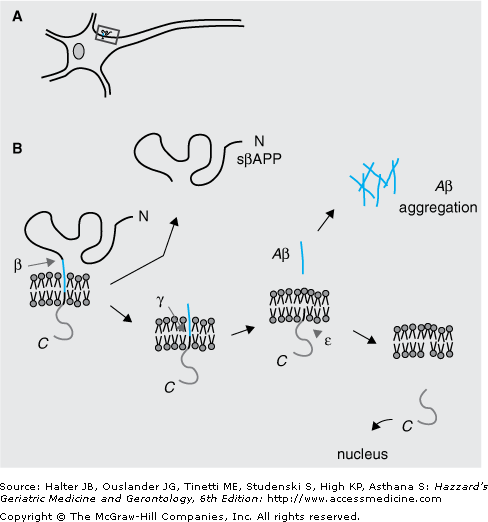
Generation of amyloid β-peptide (Aβ) from amyloid precursor protein (APP). APP is a type 1 membrane protein with a large extracellular domain, a single membrane-spanning domain, and a short cytoplasmic tail. The Aβ region of APP (in yellow) includes the first 12 to 14 amino acids of the membrane domain. (A) Shows a schematic image of APP on the cell-surface of a neuron, whereas (B) provides a closer view of APP processing. The initial enzymatic step for the generation of Aβ requires proteolysis of APP at β site (amino acid 1 of the Aβ region). This event liberates a large N-terminal fragment (sβAPP) that is rapidly secreted into the extracellular milieu and a small C-terminal fragment (β-APP-CTF) of 99 amino acids (also called C99). The removal of sβAPP most likely induces a conformational change that allows subsequent cleavage by γ-secretase. Once generated, the Aβ peptides aggregate in the brain in the form of plaques. Further cleavage of β-APP-CTF at the site liberates the signaling active APP intracellular domain (AICD). In addition to the above β/γ pathway, APP can also be cleaved at the α site (between amino acids 16 and 17 of the Aβ region) precluding the generation of Aβ.
The molecular mechanisms underlying the toxicity of Aβ are still being investigated and currently incompletely understood. However, recent research seems to indicate that small Aβ aggregates (oligomers), which represent the “preplaque” neurotoxic species of Aβ, act as the proximate cause of neuronal injury and synaptic loss associated with AD. Additionally, the C-terminal tail of APP can undergo further processing at amino acid 664 of APP695 liberating two small cytosolic fragments, Jcasp (from aa. 649 to 664 of APP695) and C31 (containing the last 31 amino acids of the C-terminus of APP, from aa. 665 to 695). Both these fragments are generated only after γ cleavage, require caspase-mediated processing of APP, and can activate proapoptotic pathways in a variety of cellular systems.
It is widely known that, although the major source of Aβ, APP exerts several important functions in the nervous system. APP comprises a group of ubiquitously expressed proteins that are generated from one single gene by differential splicing, resulting in three major isoforms of 695, 751, and 770 residues. The first isoform (APP695) is highly abundant in neurons and differs from the others in the absence of a 56-amino-acid domain that is homologous to the Kunitz-type of serine protease inhibitors. Among others, some of the important proposed normal functions of APP include serving as a cell-surface receptor, growth factor, protease inhibitor, cell–cell interaction molecule, coreceptor/partner in the endocytic/lysosomal network, coagulation inhibitor factor, cell-surface scaffold protein, kinesin-interacting molecule for axonal transport, and transcription factor.
The most critical clinical link between Aβ and AD came from the observation that patients with Downs’ syndrome (trisomy 21) had a higher propensity of developing a clinical and pathological phenotype resembling AD, thereby suggesting a potential association between AD and chromosome 21. This observation was further strengthened by the fact that Aβ was the major component in plaques from both patients with Downs’ syndrome and AD, and that its genesis was related to a gene (APP) located on chromosome 21, close to the obligate Downs’ syndrome region. Following the identification of APP, several groups found mutations in the APP gene that were linked to familial forms of AD. Given that the duplication of the APP locus could result in early AD and that Downs’ syndrome patients with partial trisomy 21 developed AD only when the trisomy was proximal to the APP locus, the potential direct relationship between APP metabolism and AD seems strong. Furthermore, causative mutations in the genes that encode for presenilin 1 (PS1) and presenilin 2 (PS2), which are also implicated in the metabolism of APP, have been found and are associated with familial forms of AD, thereby conferring additional strength to the linkage between APP/Aβ metabolism and AD.
Although the generation of Aβ from APP seems to be a pivotal step in the pathobiology of AD, it does not explain all the neuropathological changes observed in patients with AD. For example, examination of the brain of transgenic mice expressing human APP harboring one or more FAD-associated mutations reveals presence of amyloid plaques and some synaptic loss and cognitive deficits, but absence of tau pathology and astrocytosis. This suggests that additional biochemical/molecular events are required to develop the full pathological spectrum of AD. To circumvent this issue, several new animal models have been generated where human APP is accompanied by additional genes. These genes include the presenilins (harboring FAD-associated mutations), tau, and ApoE. Recently, several transgenic mice models harboring three or five FAD-associated mutations (respectively called 3X and 5X mice) in two or more genes have been generated. All of these models demonstrated that Aβ is an essential element for the development of AD-like neuropathology and revealed a close relationship between Aβ and the phosphorylation/aggregation state of tau. However, none of the mouse models fully reproduce the classical AD-phenotype; thereby again suggesting that Aβ seems to be necessary but not sufficient to produce the entire spectrum of AD neuropathology.
Transgenic mice expressing the human microtubule-associated protein tau develop the typical tau-related pathology found in individuals suffering from frontotemporal dementia with parkinsonism; however, they do not develop amyloid plaques suggesting that tau is not required for the formation of plaques. Crossing these mice with APP transgenic mice potentiates tau-related pathology and neuronal loss but does not aggravate plaque pathology suggesting that Aβ acts upstream of tau in the classical AD-phenotype. However, studies from patients with AD, mouse models, and ex vivo cellular systems indicate that Aβ and tau can interact synergistically, thereby fostering their respective aggregation and neuronal loss. The true relationship between Aβ and tau is more complex than previously thought and likely involves additional molecular and biochemical pathways acting upstream of both Aβ and tau in the AD brain.
The recent identification of the gene that encodes BACE1, the rate-limiting enzyme in the biosynthesis of Aβ, prompted the generation of “knock-out” mice that lack BACE1. Crossing these mice with the APP mutant mice models revealed that elimination of β cleavage of APP is sufficient to resolve many aspects of AD pathology, including the synaptic impairment and cognitive phenotype of the animals. Moreover, these approaches indicated that BACE1 represents a viable target for development of effective treatment strategies for AD, given that disruption of BACE1 does not result in a strong phenotype in the mouse. Overall, there is extensive evidence from laboratory research that Aβ plays a major role in the pathogenesis of AD; however, there are likely additional, yet unidentified, molecular pathways that contribute to the overall pathobiology of AD.
Epidemiology and Risk Factors for Alzheimer’s Disease
Alzheimer’s disease is the most common cause of dementia in older adults, currently affecting more than five million Americans and more than 24 million individuals around the world. Unless effective preventive strategies are identified, it is anticipated that the prevalence of AD will double every 20 years. Of note, the United Nations predicts that the major rate of increase in the prevalence of AD will likely occur in developing countries like India and China that may not possess all the essential resources, public health support system, or medical expertise to care for patients with AD.
There is clear evidence that a number of risk factors significantly enhance the overall risk for developing AD. These risk factors relate to both genetic and nongenetic markers, and are discussed below.
Aging is the single most important and validated risk factor for AD. The projected increase in AD prevalence in the coming decades is in part related to recent increases in the average human life span. Almost all of the epidemiological studies indicate that the incidence of AD increases with aging. For example, in the year 2000, the age-specific distribution of patients with AD was 5% between the ages of 65 and 74 years, 18% between the ages of 75 and 84 years, and 45% above the age of 85 years. Additionally, age-associated increases in the amount of amyloid plaques and NFTs in the brains of older persons further underscore a strong relationship between aging and AD pathology. Although not clearly understood, converging findings from recent research provides some clues concerning the potential molecular pathway(s) underlying the association between aging and AD. This pathway acts downstream of the insulin-like growth factor 1 receptor (IGF1-R) and plays a major role in determining both the lifespan and age-associated diseases. In general, activation of IGF1-R signaling accelerates aging and shortens lifespan, while a partial block—as achieved with either genetic or biochemical approaches—delays aging, extends lifespan, and, most importantly, delays the progression of many age-associated diseases, including AD and type 2 diabetes mellitus. Recent biochemical and genetic studies have delineated a precise molecular pathway involving IGF1-R, neurotrophin signaling, BACE1, and APP metabolism, suggesting that pharmacological approaches aimed at IGF1-R signaling might help delay the progression as well as onset of AD.
Aging and IGF1-R signaling are both associated with cerebrovascular dysfunction, which, emerging evidence supports, may play a key role in the development of AD. An increased exposure time to age-dependent vascular risk factors or an interaction between aging and vascular risk factors may in part account for the effects of aging on the pathobiology of AD. Thus, targeting treatment of vascular risk factors may ameliorate some of the age-associated risk for AD.
Late-onset AD is the most common form of the disorder, accounting for more than 90% of all AD cases. Although early-onset AD has strong genetic links, many cases of LOAD are seen in individuals without any clear genetic predisposition. A common polymorphism in the apolipoprotein E (APOE) gene is the major determinant of risk in families with LOAD. Of the three common allelic forms (APOE2, APOE3, and APOE4), APOE4 is associated with up to fourfold increased risk of developing AD among individuals with at least one APOE4 allele. However, APOE4 genotype is neither necessary nor sufficient for the development of AD. APOE4 allele may contribute to AD by influencing processes related to the development of AD, including altering the rate of production, clearance, or aggregation of amyloid β-peptide and/or influencing cerebral cholesterol metabolism and inflammation. Currently, routine genetic testing for LOAD is not recommended. Recent estimates based on twin studies suggest that nearly 50% of LOAD cases may be related to APOE allele and other unidentified genetic factors.
Although controversial, there is some evidence that AD is more common among women. Specifically in the population-based studies published to date, more than half reported a greater risk of AD in women, while the others found no difference. It is generally believed that the discrepant findings between various studies are likely owing to several important reasons, including methodological differences and a failure to account for the potential gender-related variability in education, occupation, and lifestyle variables that can directly affect the risk of AD. Some data support that estrogen deficiency following menopause may contribute to the development of AD; however, the effects of estrogen replacement therapy on cognition remains controversial.
There is mounting evidence that midlife vascular risk factors are associated with an increased risk of AD in later life. In epidemiological studies, hypercholesterolemia, hypertension, hyperhomocysteinemia, obesity, diabetes mellitus, and elevated inflammatory markers have all been associated with enhanced risk of developing AD. Furthermore, APOE4 allele is related to abnormal cholesterol metabolism suggesting that the increased risk for AD associated with this genotype may be partially mediated through dysregulation of cholesterol. The cholesterol–AD connection is further supported by results from animal studies showing that hypercholesterolemia stimulates the amyloidogenic pathway of APP leading to increased production and deposition of Aβ. Conversely, decreased levels of cholesterol produce the opposite effect reducing the production and deposition of Aβ. Not only do some vascular risk factors appear to influence Aβ metabolism, but conversely, soluble Aβ has been shown to induce dysfunction in cerebral vessels of rats and in cultured endothelial cells. As both Aβ deposition and cerebrovascular dysregulation are two early findings in preclinical AD pathology, these processes most likely work synergistically to accelerate neuronal degeneration. Although the evidence supporting an association between vascular disease and AD is increasing, it is presently unclear whether treating these modifiable vascular risk factors will affect the development or progression of AD. Clinical trials are currently underway to evaluate the potential role of treating these risk factors in preventing or delaying AD progression.
The fact that cerebrovascular diseases such as vascular dementia and cerebral amyloid angiopathy (CAA) share common risk factors with AD supports that their pathologic mechanisms are related. A clear separation between vascular forms of dementia and classical AD based on cognitive impairment is extremely difficult. Typical patients with vascular dementia have a history of vascular disease and show a more pronounced impairment in executive function with only mild memory deficits. The presence of white matter hyperintensities and lacunar infarcts demonstrated by MRI constitutes an important criterion to support the diagnosis of vascular dementia. However, only 2% to 3% of patients with dementia fulfill the diagnostic criteria of pure vascular dementia. In contrast, a large group of patients show an overlapping phenotype with subcortical white matter changes, hypoperfusion, microinfarcts, and amyloid angiopathy. These patients tend to progress into classical AD and will show some degree of amyloid plaques and tau aggregates at autopsy, together with microglia and astrocytic proliferation. CAA is characterized by abnormal accumulation of fibrillar Aβ (especially Aβ40) in the walls of meningeal and cortical arterioles, venules, and capillaries. The damaged vessels undergo spontaneous rupture, and the patients often present with subcortical white matter changes and microhemorrhages. An autosomal dominant form of CAA is caused by a point mutation in the APP gene (Dutch-type) and is a form of FAD. Understanding the overlapping pathologic mechanisms among AD, vascular dementia, and CAA may help to identify common targets for disease-modifying therapies.
Several epidemiological studies have indicated that a history of severe or repeated head trauma is a risk factor for cognitive decline and AD. The majority of patients develop AD, although a minority might manifest dementia of Parkinson disease and depression. Neuropathological examination of the brain from patients with a history of head trauma generally reveals changes of diffuse amyloid plaques together with tau pathology, inflammatory response, and loss of cholinergic neurons. A smaller number of patients show diffuse α-synuclein reactivity, Lewy body–type aggregates, and loss of dopaminergic neurons resembling Parkinson disease. In animals, pathological changes following experimental head trauma reveal transient upregulation of BACE1 together with increased generation of Aβ. These features are accompanied by tau hyperphosphorylation and increased caspase-mediated cleavage of APP. Thus, head trauma may lead to AD by triggering accelerated neurodegeneration.
Depression is a common psychiatric condition in the elderly and widely reported to present with symptoms of cognitive and functional impairment, commonly grouped into the clinical entity of pseudodementia. Furthermore, more than 30% of patients with AD develop depression during the course of their illness, and might present with the symptoms of depression as the first clinical manifestation of underlying AD. Thus, a better understanding of the potential relationship between depression and AD is of pivotal clinical significance. Although not confirmed universally, there is converging evidence from several studies that a history of depression can increase the risk of AD later in life. Findings of a recent meta-analysis and metaregression analysis involving 20 retrospective and prospective studies supported an increased risk of AD in patients with prior history of depression. The pooled odds ratio for developing AD in this analysis was 2.03 for case–control and 1.90 for cohort studies. To date, the precise mechanism(s) underlying the association between depression and enhanced risk for AD is unknown. However, several potential mechanisms have been proposed, including elevated levels of cytokines observed in both patients with AD and depression, increased association of vascular risk factors with the pathobiology of AD as well as depression, and the potential role of APOE4 allele in enhanced risk for both AD and depression. Clearly, more research is necessary to better understand the biological basis of increased risk of AD in patients with a history of depression.
There is some evidence, albeit controversial, from epidemiological studies that higher education may protect against the development of AD. Individuals with elementary level education might be at an increased risk of developing AD, compared to those with high school and college education. Of note, these findings have not been substantiated by all studies and need to be systematically evaluated in larger epidemiological studies. The exact mechanism underlying the neuroprotective effects of higher education is currently unknown; however, based on the “use it or lose it” theory there is suggestion that individuals with higher education might have more neurons to lose before they cross the threshold of developing dementia.
It is generally presumed that AD affects all ethnic groups equally. However, there is some recent evidence that the disease might be more prevalent in certain ethnic groups. In a population based study in the Washington Heights and Inwood communities of New York City, the cumulative incidence of AD was increased twofold among individuals of African-American and Caribbean Hispanic origin. This ethnicity-based incidence of AD did not change following corrections for differences in the number of years of education, or a history of diseases like hypertension, diabetes mellitus, and cardiovascular disease. Although not universally confirmed, other studies also support these findings. In a study in Houston, both the incidence and prevalence of AD were higher among African-Americans and Hispanics than retirees of Caucasian origin. Although speculative, it is possible that the increased risk of AD among the above ethnic groups may be because of a higher incidence of comorbid vascular and cardiovascular diseases and a higher prevalence of APOE4 genotype.
Clinical Presentations of Alzheimer’s Disease
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


