Hypogonadotropic hypogonadism (low LH and FSH)
Constitutional delay of growth and puberty (CDGP)
Functional hypogonadotropic hypogonadism: Delayed, but spontaneous, pubertal development
Permanent idiopathic hypogonadotropic hypogonadism (IHH)
Hypothalamic and pituitary dysfunction
CNS tumors: Germinoma, optic glioma, oligodendroglioma, Rathke’s pouch/cleft cyst astrocytoma, pituitary tumor
Panhypopituitarism
Isolated gonadotropin deficiency
Hypophysitis
Langerhans histiocytosis
Radiation therapy
Head trauma
Congenital malformation with midline central defect (Septo optic dysplasia)
Mutations in the PROP1, LHX3, and HESX1 genes
Syndromes: Prader–Willi syndrome, Coffin–Lowry syndrome , CHARGE syndrome, Laurence–Moon–Biedl syndrome, and others
Mutations in the NR0B1, GPR54 genes, GnRH receptor gene mutations, inactivating mutations of KISS 1 and KISS 1 R genes, loss-of-function mutations in genes encoding neurokinin B and its receptor, DAX-1 mutations
Hypergonadotropic hypogonadism (increased LH and FSH)
Gonadotropin receptor mutations
FSH β subunit gene mutation
LH/FSH receptor mutation
Gonadal dysgenesis
Turner syndrome
Premature ovarian failure
Resistant ovary syndromes
Irradiation
Cytotoxic therapy
Trauma
Infections
Galactosemia
Glycoprotein syndrome type1
AIS with gonadectomy
Androgen resistance
Eugonadotropic hypogonadism (normal LH and FSH)
Steroidogenic enzyme defects
Cholesterol desmolase complex deficiency (lipoid adrenal hyperplasia)
3-β OH-steroid dehydrogenase deficiency
17 α-hydroxylase deficiency
C17,20-desmolase deficiency
17-β OH steroid oxidoreductase deficiency
21-hydroxylase deficiency in girls
Anatomic abnormalities
Imperforate hymen
Vaginal atresia
Vaginal and uterine agenesis (Mayer Rokitansky–Kuster–Hauser syndrome)
PCOS
Prolactinoma
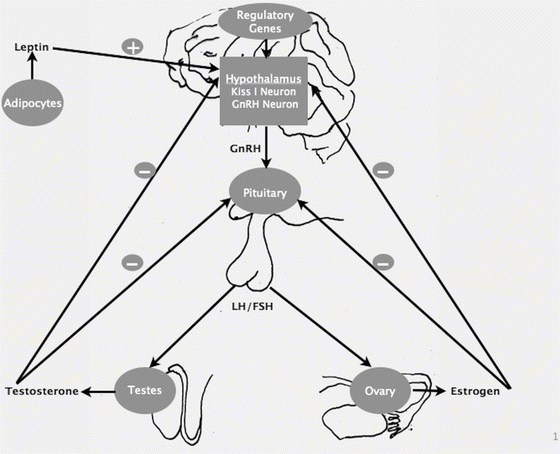
Fig. 17.1
Schematic illustration of pubertal regulation, demonstrating the factors and hormones providing positive and negative feedback control
Introduction
Puberty is initiated when the hypothalamic gonadotropin-releasing hormone (GnRH) pulse generator begins secreting brief nocturnal pulses of GnRH from the hypothalamic arcuate nucleus that subsequently stimulate the pituitary to release luteinizing hormone (LH) and follicle-stimulating hormone (FSH) [2]. A recently discovered hormone, kisspeptin, acts on the hypothalamic GnRH neurons, stimulating GnRH secretion [3]. The gonadotropins (LH and FSH) promote gonadal maturation and gonads synthesize sex steroids, including testosterone and estrogen, and other proteins. LH acts on theca cells and interstitial cells to produce progestins and androgens which diffuse into adjacent granulosa cells. FSH acts on granulosa cells to stimulate aromatization of these androgens to estrogen. Estrogen and testosterone then promote pubertal changes throughout the body and provide negative feedback effect on the GnRH and gonadotropins (Fig. 17.1). The first physical signs of puberty are typically breast development in girls and testicular enlargement in boys (testicular volume >3 ml/≥2.5 cm in length). Some children, especially girls, have the appearance of pubic hair prior to the initiation of breast development, but in the absence of other puberty signs this usually represents adrenarche [adrenal source of androgens, independent of hypothalamic–pituitary–gonadal (HPG) axis maturation] and not true puberty. The trigger(s) for reactivation of the HPG axis is not completely understood; but, modifying factors include general health, nutrition, genetic determinants, and pubertal timing among primary relatives. Elevated body mass index is associated with delayed puberty in boys [4, 5]. Many of the genes involved in the HPG axis maturation are still unknown. Kisspeptin-1 and its cognate receptor (GPR54, a G-protein-coupled receptor) are integral to the normal function of HPG axis and play a critical role in the physiologic regulation of puberty [3, 6]. Kisspeptin is co-expressed with neurokinin B and dynorphin and hence these signaling pathways are also important in physiologic regulation of puberty [7]. There is evidence that leptin, a 16 kDa hormone product of the Ob gene, synthesized by adipocytes, plays a permissive role [8, 9].
Although the lower limit of normal for the onset of puberty is contestible, the average age for this process is generally accepted to be 9–10 years for girls and 10–11 years for boys [2, 10]. Delayed puberty can be defined as failure to demonstrate signs of pubertal maturation by an age that is ≥2 standard deviations above the population mean [2]. Lack of testicular enlargement by age 14 in males, lack of breast development by age 13 in females, absence of menarche by age 16 in girls, or absence of menarche within 5 years of pubertal onset [11, 12] is defined as, delayed puberty. Interestingly, males present far more often for evaluation of delayed puberty, but it has been suggested that this is in part due to a referral bias [1, 13].
Hypogonadotropic Hypogonadism
Hypogonadotropic hypogonadism (HH) is the commonest of the groups in both sexes and includes many pathologic disorders that can be further subdivided into constitutional delay of growth and puberty (CDGP), functional hypogonadotropic hypogonadism (FHH), and permanent hypogonadotropic hypogonadism (PHH). This classification can facilitate the diagnostic process by appropriately directing early evaluation efforts. HH is defined as lack of normal gonadal function secondary to low or absent gonadotropin function. In this case, the problem can be in the pituitary gland itself, or it can be related to hypothalamic dysfunction (delayed activation of the gonadotropin-releasing hormone (GnRH) pulse generator). In addition to low or absent LH/FSH, sex steroid concentrations will be in the prepubertal range and bone age is typically delayed. Concentrations of adrenal androgens may be normal. The delay in puberty can be either temporary or permanent. Isolated hypogonadotropic hypogonadism is diagnosed if endogenous puberty has not begun by the age of 18 years [2].
Constitutional Delay of Growth and Puberty
CDGP is the most common cause of delayed puberty especially in males (65 % of boys and 30 % of girls with delayed puberty) [1]. It is a benign variant of normal growth and development and, is notably, a diagnosis of exclusion. Typically, a child will be of normal size at birth and in infancy. At some point in early childhood, a decrease in growth velocity (GV) causes a decline in height centile for age growth curve. Normal growth then resumes with the child growing at an age-appropriate GV and at a consistent but low height percentile for age. This represents a global delay in biologic maturity affecting puberty and bone maturation. Height is usually appropriate for genetic potential when plotted for bone age. There is also usually a family history of “late bloomers” in the family—history of delayed puberty in the patient’s parents or siblings (77 %) [14]. They exhibit a relatively normal prepubertal growth velocity and protracted prepubertal growth nadir. The discordance in height vs. peers is exacerbated by a relatively lower growth velocity compared to peers who are experiencing a pubertal growth spurt. The growth velocity and height, however, should remain normal for bone age and pubertal stage. After HPG axis maturation, secondary sexual characteristics appear in their natural sequence with normal secondary sexual characteristic development. In 93 % of cases spontaneous pubertal maturation occurs by 18 years and has an excellent outcome. In some cases, the constitutional delay of puberty superimposed on constitutional short stature and final height may be shorter than genetic potential [15].
Diagnosis of this condition is ultimately a matter of watchful waiting with close monitoring of growth and development. However, judicious evaluation to rule out other conditions and to support the likelihood of CDGP is important. Clustering of CDGP has been clearly established [1, 14] and the pericentromeric region of chromosome 2 harbors a gene predisposing to pubertal delay [14]. Growth charts, if available, should be reviewed to demonstrate typical CDGP pattern. In CDGP, both adrenarche and gonadal enlargement occur later than average; whereas in isolated HH, there is dissociation of adenarche and gonadarche, with adrenarche occurring at normal age [1, 16]. GV and serum somatomedin-c (IGF1) should be monitored and remain normal for pubertal status. Evaluation may reveal low/normal gonadotropins and a delayed bone age. However GnRH agonist stimulation testing is not helpful in differentiating CDGP and permanent HH [17]. A positive response to GnRH agonist is suggestive of CDGP. To date, no single lab test or hormone stimulation protocol has the sensitivity and specificity to make this diagnosis. Response to sex steroid replacement therapy, which will often trigger activation of the hypothalamic–pituitary–gonadal (HPG) axis in CDGP but not in permanent HH, may aid in the diagnosis. Baseline, morning testosterone concentration of ≥20 ng/dl suggests the appearance of pubertal signs within 12–15 months [18]. A very low basal serum follicle-stimulating hormone (FSH) [<0.2 IU/L by immunochemiluminometric assays (ICMA) and <1.0 U/L by immunofluorometric assays (IFMA)] is suspicious of HH [19, 20]. Serum inhibin B (INHB) measurement will help to discriminate HH from CDGP. INHB is produced by sertoli cells upon FSH stimulation and is a reflection of sertoli cell integrity [21]. A baseline INHB concentration of >35 pg/ml is highly suggestive of CDGP [21].
Functional Hypogonadotropic Hypogonadism
FHH represents another form of temporary, reversible HH. It accounts for about 20 % of children with delayed puberty [1]. Within this category is a broad range of pathology that highlights the complexity of the HPG axis and the diverse factors that must coordinate to initiate puberty. The most common diagnoses are related to chronic or underlying illnesses, such as hypothyroidism, cystic fibrosis, Crohn’s disease, inflammatory disorders that produce cytokines, immunosuppression seen in perinatally HIV-infected children, and chronic renal failure [1, 22, 23]. The mechanism of pubertal delay in the case of underlying illness is thought to be manifold, involving a combination of factors that include, but not limited to, undernutrition, stress, and medications such as corticosteroids resulting in abnormal gonadotropin secretion [22]. The implicated genetic variations are in genes that have been associated with idiopathic hypogonadotropic hypogonadism [24]. As aforementioned, a common cause of FHH is malnutrition, as seen in anorexia nervosa or intense exercise resulting in HPG dysfunction. The connection between weight, especially body fat mass and puberty, has been extensively studied [8, 25, 26].
A thorough and detailed history may reveal systemic complaints, eccentric eating habits, or an obsession with exercise and weight loss. Physical exam may be revealing at times: weight and BMI will typically be low for age, and erosion of dental enamel and callused knuckles may suggest eating disorders. Laboratory evaluation may demonstrate elevated sedimentation rate and/or other inflammatory markers. Further evaluation depends on the clinical situation, but thyroid function should be assessed in all cases of HH.
Isolated Gonadotropin Deficiency
Isolated GnRH deficiency resulting in low or inappropriately normal gonadotropins and absent or incomplete puberty could be associated with abnormalities in craniofacial, skeletal, neurologic, renal, and olfactory systems. X-linked gene, KAL1, found in GnRH-deficient men causes isolated gonadotropin deficiency known as Kallman’s syndrome (KS) [27, 28]. This condition is caused by abnormal migration of embryonic GnRH and olfactory neuronal cells to the hypothalamus, resulting in HH and anosmia or hyposmia. FGFR1 mutations are autosomal and are often associated with cleft lip/cleft palate, syndactyly, or skeletal abnormalities. Other cases of permanent isolated HH have historically been referred to as idiopathic HH (IHH). More recently, several genetic mutations have been discovered in some of these cases. Rare sequence variants (RSVs) in genes involved in GnRH neuronal migration (FGF8, FGFR1, KAL1, PROK2, PROKR2, and NELF), secretion (GNRH1, GPR54, TAC3, and TACR3), and receptivity (GNRHR) have been reported to contribute to GnRH deficiency in both men and women [28, 29]. Most notably GnRH receptor mutations causing GnRH insensitivity and G-protein-coupled receptor 54 mutations causing impaired gonadotropin secretion have been identified [27]. At the time of puberty, the affected patients may have adrenarche—some pubic hair may be there, but little or no breast development or axillary hair and present with primary amenorrhea. HH is reported in leptin deficiency, where puberty can be induced by recombinant leptin [9]. HH has also been reported in DAX1 mutations. DAX1 is a nuclear receptor protein encoded by the NR0B1 and associated with X-linked congenital adrenal hypoplasia and HH, resulting from defects in the production of gonadotropins by the pituitary.
In all cases of isolated HH, other pituitary hormones should be assessed to confirm that the defect is truly isolated to gonadotropin secretion. In KS there is often associated decreased olfaction, synkinesia (mirror movements), sensorineural deafness, unilateral renal agenesis, and pes cavus. Brain imaging in cases of KS may show aplasia/hypoplasia of olfactory bulb and sulci. Some patients with isolated HH will have a positive family history, but most cases are sporadic. Genetic testing for associated mutations is possible, but not sensitive for diagnosis as the majority of isolated HH is idiopathic that is not associated with identified genetic abnormalities [3]. It can be particularly challenging to differentiate between CD and isolated HH. A reversible form of congenital GnRH deficiency also has been identified where the activation of the GnRH–gonadotropin axis is markedly delayed and the affected subject undergoes a sustained reversal of hypogonadotropism by age 18 [30]. Definitive diagnosis of GnRH deficiency cannot be made before 18 years.
Multiple Pituitary Hormone Deficiencies
Hypogonadotropic hypogonadism as part of a constellation of multiple pituitary hormone deficiencies (MPHDs) can occur in the setting of central nervous system (CNS) tumors (i.e., craniopharyngioma, germinoma, hypothalamic glioma, prolactinoma), non-tumoral lesions (i.e., histiocytosis, granuloma, hydrocephalus, vascular lesions), cerebral dysgenesis, CNS trauma or infection, and destructive medical therapies such as radiation therapy. Genetic mutations, including defects in transcription factors such as PROP1, HESX1, LHX3, and LHX4, have also been identified in these cases [27].
Craniopharyngiomas are the predominant cause of permanent HH in children [1]. They are benign tumors that arise in the suprasellar region of the brain and may cause symptoms related to increased intracranial pressure and/or pituitary gland and optic nerve dysfunction. Growth hormone deficiency is the most common endocrinologic disorder, but all pituitary hormones can be affected and most adolescents presenting with these tumors will have delay in puberty [31]. Surgery and radiation therapy may further damage pituitary and hypothalamic function leading to permanent hormone deficiencies. Depending on dose and anatomical location, intracranial radiation therapy, in particular, causes irreversible damage to the hypothalamic–pituitary axis. It usually affects the hypothalamus to a greater extent than the pituitary gland and precocious puberty is more common than delayed puberty [32].
Septo-optic dysplasia (SOD) with midline cerebral dysgenesis can cause pubertal delay. It is characterized by congenital absence of the septum pellucidum, bilateral optic nerve hypoplasia, and hypopituitarism. There is significant variability in the severity of affected children, but typically involves visual impairment and pituitary hormone deficiency with radiologic abnormalities of the septum pellucidum or corpus callosum. It is occasionally associated with HESX1 gene mutations [33].
In any case of MPHD, laboratory assessment of thyroid function, adrenal function, growth, and electrolyte balance is indicated. Physical exam should include a thorough neurologic examination. A careful history should include review of past or recent head trauma, CNS infection, or intracranial radiation therapy. A review of systems should be performed with particular attention to visual change, headache, vomiting, fever, polyuria, polydipsia, poor growth, and salt craving. In most cases of MPHD, brain and pituitary imaging is a requisite and, if SOD is considered, an ophthalmologic exam is indicated to evaluate optic nerves and vision. Genetic testing is not indicated in all patients, but for those with a family history of MPHD and specific radiographic findings, targeted testing for specific mutations may be indicated [34].
Genetic Syndromes
There are several congenital syndromes that have HH as one of the primary findings. The most well-known syndrome is Prader–Willi syndrome (PWS). It is caused by loss of imprinted genetic material from the paternally derived chromosome 15. In addition to HH, it is marked by neonatal hypotonia, feeding problems in infancy, obesity, hyperphagia, developmental delay, small hands/feet, and short stature. It is usually sporadic.
CHARGE syndrome is another common syndromic cause of HH [1]. This acronym stands for coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear abnormalities, and/or hearing loss. Mutations in the CHD7 gene have recently been identified in around 2/3 of affected patients [35] and it has been suggested that the developmental abnormality causing HH in this condition may be similar to that seen in KS [36]. It is an autosomal dominant mutation but most cases are sporadic. Bardet–Biedl syndrome also includes HH as a primary feature. Other manifestations of this rare, autosomal recessive condition include rod-cone dystrophy, obesity, renal dysfunction, developmental delay, and postaxial polydactyly.
The presence of other dysmorphic characteristics associated with a syndrome in addition to HH warrants further evaluation, e.g., for PWS, genetic testing, preferably with DNA-based methylation testing [37]. CHARGE and Bardet–Biedl syndromes are both diagnosed clinically. CHARGE syndrome diagnosis is based on major and minor criteria, but genetic testing for CHD7 mutation is available. Similarly, there is genetic testing for 14 associated genetic mutations for Bardet–Biedl, but the diagnosis is based on the presence of primary and secondary phenotypic features [38].
Hypergonadotropic Hypogonadism
Hypergonadotropic hypogonadism (HHG) causes delayed puberty due to primary gonadal failure. By definition, these disorders have elevated levels of gonadotropins without concomitant increase in sex steroid concentrations and without signs of pubertal maturation. Within this category lie primarily disorders of gonadal dysgenesis and gonadal injury.
Gonadal Dysgenesis
Gonadal dysgenesis is the most common cause of HHG in children [1]. It is usually related to chromosomal abnormalities and hence chromosomal analysis is fundamental in the evaluation of children with HHG. In females, Turner’s syndrome (TS) is a condition of X-monosomy (45, X) or structural abnormalities of an X chromosome. Mosaicism is common (50 % may have 45X/mosaic karyotype). Girls have short stature and lack of normal pubertal development caused by streak ovaries and premature ovarian failure. The degree of pubertal maturation is variable with occasional spontaneous menarche and rare fertility [39]. Other characteristics include heart and renal abnormalities, webbed neck, and broad chest. Mixed gonadal dysgenesis (MGD) can also occur similarly with X-monosomy/XY mosaicism. This protean genetic disorder can range in phenotypic presentation depending on the degree of mosaicism, from phenotypic female to phenotypic male.
Klinefelter’s syndrome is a chromosomal abnormality found in males presenting with delayed puberty. In this case the underlying karyotype is 46-XXY. Along with HHG, this condition is characterized by tall stature, gynecomastia, decreased upper to lower segment body ratio, and learning disabilities. Other less prevalent disorders of gonadal dysgenesis in 46XY karyotype are Swyer syndrome (46XY, streak gonads), Drash syndrome, Frasier syndrome, mutations of SOX-9, DAX 1 with duplication of Xp21, and mutations in the SF 1 [12]. Additionally, certain disorders of sex development can present as HHG. For example, children with AIS or 5-alpha reductase deficiency (5-ARD) are genetically XY but are often raised in female because of ambiguous or female external genitalia. They may present with pubertal delay or primary amenorrhea when there is a failure to progress through normal female puberty.
Rare cases of gonadotropin receptor mutations (LH/FSH receptor mutation in XX females) with normal breast development, primary or secondary amenorrhea, elevated serum LH/FSH [depending on mutation of LH/FSH receptor], low estradiol level, and infertility have been reported. LH receptor mutation (homozygous or compound heterozygous inactivating mutations of the LH receptor) in XY males presents with male pseudohermaphroditism—female external genitalia/micropenis, absence of Mullerian structures, Leydig cell hypoplasia, lack of breast development, and HHG [11, 40, 41].
FSH β subunit gene mutation presents with delayed puberty, primary amenorrhea, elevated LH, and low or undetectable FSH [42, 43]. FSH receptor mutation presents with primary gonadal failure and HHG in females [42]. FSH is required for follicular development and ovarian androgen and estrogen synthesis in females. Males present with oligospermia, but are fertile as FSH is not necessary for spermatogenesis [43].
Gonadal Injury or Loss
HHG also occurs in children who have suffered gonadal damage, frequently as a result of treatment for an underlying malignancy. Gonadal tissue is particularly sensitive to radiation damage, but can also be affected by many chemotherapeutic agents [1]. Testicular tissue is more sensitive to damage by these cytotoxic therapies compared to ovarian tissue, and in all cases the risk is agent and dose dependent [44].
Gonadal tissue can also be injured by a wide spectrum of other processes, including trauma, infarction, and infection. In addition, certain disease processes can affect gonadal tissue and lead to pubertal dysfunction and infertility. Autoimmune polyendocrine syndrome type 1 (APS 1), for example, is associated with autoimmune-induced damage to gonadal tissue. Gonadal failure is much more common in females with this disorder and there is correlation between SCC autoantibodies and ovarian failure in women with APS 1 [45]. Galactosemia is also associated with HHG in female patients, especially those for whom treatment was delayed. It is thought that this is caused by cellular galactose toxicity occurring very early in life [46].
Complete loss of gonadal tissue can also present with delayed puberty. There are several indications for gonadectomy in the prevention and treatment of malignancy, including mixed gonadal dysgenesis and selective cases of androgen insensitivity syndrome (AIS) [47]. Additionally, anorchia is a male condition in which testes form normally in utero, as evidenced by normal male genitalia, but are absent at the time of birth, indicating loss sometime after the 14th-week gestational age. Cause is unknown. “Resistant ovary syndrome” is a condition due to abnormalities in gonadotropin receptors or antibodies to these receptors seen in 46 XX karyotypes, typically presenting with sexual immaturity and primary amenorrhea, small ovaries with primordial follicles despite elevated gonadotropin concentrations [48, 49].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


