Fig. 9.1
Diagnostic workup in Cushing’s syndrome. ACTH adrenocorticotropin, BIPSS bilateral inferior petrosal sinus sampling, CRH corticotropin releasing hormone, CD cushing’s disease, CT computed tomography, ECS ectopic Cushing’s syndrome, LDDST low-dose dexamethasone test, NSaC night salivary cortisol, MRI magnetic resonance imaging, MS metabolic syndrome, MNC midnight cortisol, PET positron emission tomography, UFC 24 h-urinary free cortisol, 1-DST 1 mg overnight dexamethasone test
The most important step for the diagnosis of CS is the clinical suspicion along with a detailed past medical and drug history [1]. A vast range of signs, symptoms and other abnormalities along with a wide range of severity can be seen in CS (Fig. 9.1). When the presentation is florid including central obesity with limb wasting and muscle weakness, a plethoric face with hirsutism and frontal balding, spontaneous bruising along with metabolic abnormalities (hypertension, prodiabetes or diabetes) and osteoporotic fractures, then the diagnosis is straightforward. Protein wasting, thin skin in the young, easy bruising, and proximal muscle weakness most reliably distinguish CS from PC. In addition, while specific clinical features may suggest one rather than another aetiology, the degree of hypercortisolaemia seems to be the major determinant of the clinical features rather than its duration [7, 9]. Severe hirsutism and virilisation strongly suggest an adrenal carcinoma [3]. A gradual onset of signs may be seen with NETs as opposed to SCLCs, which usually have a more rapid onset with symptoms of profound weakness, hyperpigmentation, little or no weight gain and an absence of overt Cushingoid features, the rapidity of onset and the severity of the syndrome have been both implicated as the cause of the more frequent presence of skin pigmentation and ankle oedema in SCLCs compared to other causes of ECS [7]. On the other hand, in ECS psychiatric disorders are more prominent in NETs than with SCLCs [7, 19].
It is obvious that the similarity of different types of CS mandate the necessity for a specific step-by-step diagnostic workup to resolve any diagnostic dilemma [1]. The rationale for the diagnostic confirmation of CS is to identify the loss of the specific mechanisms controlling the tightly-controlled hypothalamo-pituitary-adrenal (HPA) axis. In normal states, the HPA axis regulates cortisol secretion from the adrenal gland under the stimulus of ACTH from the pituitary, which in turn is secreted in response to CRH and vasopressin from the hypothalamus. Cortisol then exerts negative feedback control on both CRH and vasopressin in the hypothalamus, and ACTH in the pituitary. Cortisol is also secreted in a circadian rhythm; levels fall during the day from a peak at 07.00–08.00 h to a nadir at around midnight: they then begin to rise again at 02.00 h. In hypercortisolaemic states, the normal cortisol feedback mechanism of the HPA axis is distorted with loss of the normal suppression to the exogenous administration of glucocorticoids, the circadian rhythm is lost, and cortisol production increases [16]. These considerations were in part the stimulus for the Endocrine Society to recently publish a clinical practice guideline for the diagnosis of CS [15]. Hypercortisolaemia must be established before any attempt at differential diagnosis, and this is a critical step since it is related to the number of the patients that will unnecessarily be involved in laborious and costly tests, or that will be misdiagnosed as being considered inappropriately healthy but yet suffering from the long-term consequences of the sustained hypercortisolism [20]. Hence, the initial biochemical tests should ideally have maximal sensitivity rather than specificity in order to identify individuals with the mild forms of this rare disease; later, more specific tests are used to exclude false positives. This is best performed by a combination of the following tests: 24-h urinary free cortisol (UFC; at least 2 measurements) as marker of increased synthesis of cortisol exceeding the binding capacity of corticosteroid binding globulin (CBG), low-dose dexamethasone suppression (LDDST, 0.5 mg every 6 h for 2 days) or 1-mg overnight dexamethasone suppression testing (1-DST) as marker of resistance of the HPA axis to glucocorticoid feedback and assessment of midnight serum cortisol (MNC) or late-night salivary cortisol (NSaC) as markers of the loss of circadian rhythmicity [15, 21, 22].
These tests may not be needed when a florid and severe disease is present with massively elevated serum cortisol at any time, or a urinary Cortisol more than 4× the upper limit of normal. In individuals with normal results in the initial investigation but with strong suspicion or when progression is documented, or when only one of test results is abnormal and clinical suspicion is low, further evaluation at a later stage is crucial to confirm or exclude the diagnosis [22]. It has been suggested that in these particularly difficult cases a dexamethasone-CRH test is mandated, but not all CS experts agree on its superiority over and above the standard LDDST [15]. Diagnostic tests based on a failure of feedback regulation were originally designed for the florid rather than the mild cases we now see, and the thresholds for serum cortisol levels have inevitably changed as the assays have become more sensitive. Hence, the conventional use of the 1-DST may still be insufficiently sensitive to detect mild cases of CS, particularly in CD; UFC assessment, even with more accurate assay techniques and in the most compliant patients, has limited sensitivity, particularly in cases of mild hypercortisolism. NSaC level should be the most sensitive indicator for CS along with MNC, but the former is clearly much more practical for screening purposes to detect rapid changes in the free biologically-active cortisol concentration [21]. The salivary cortisol (SC) test may be a good substitute of serum cortisol in some test such as 1-DST, LDDST or CRH tests to differentiate CS from healthy subjects, but may not be as good to differentiate CS from PC [22]. Recently, desmopressin (DDAVP) test has been suggested as better distinguishing even mild forms of CD from PC compared to UFC, 1-DST and MNC. Overall, it is apparent from recent studies that the 3 commonly performed initial diagnostic tests for CS (NSaC, UFC and 1-DST) are complementary, and their diagnostic performance may increase by their combination [21]. In specific cases some tests may be superior to others. In renal failure SC post-1-DST is superior to UFC [23] as is the case of patients with cyclic CD (cyCD) [24], where SC would be most convenient to be performed immediately upon the “cycling-in” of CS, or SC testing in children to differentiate CS from obesity as a “friendlier” diagnostic tool for the paediatric population [25]. In patients with mild or cyCS, any of these tests may yield normal results and be misleading. The UFC in particular, even when measured on repeated occasions, cannot always exclude CS. In our opinion, because of the high sensitivity and ease with which repeated measurements can be performed, NSaC appears to be the most useful screening test [26]. As opposed to this, NSaC and UFC have been both found of limited clinical value compared to the 1-DST in the diagnosis of mild CS such as the case of subclinical cortisol-secreting adenomas (SCSA) [27–29]. It has been suggested that one can exclude CS by UFC assessment after 3 normal collections, while values 4-fold greater than the upper limit of normal can be considered diagnostic for CS [1]. However, small increases in cortisol production at the circadian nadir may not be detected as an increase in UFC as most of the cortisol secreted during any 24-h period is generally between 04.00 and 16.00 h [30, 31] but they might have a small but significant increase in night time cortisol secretion. Hence, the recent guidelines for the diagnosis of CS suggest use of the 1-DST or MNC, rather than UFC in patients suspected of having mild CS because of an adrenal incidentaloma [15].
The second step in the diagnostic cascade is to establish the cause of CS; plasma ACTH measurement will be either very low indicating an adrenal cause causing ACTH suppression or readily detectable. A plasma ACTH >20 ng/L will immediately establish ACTH-dependence, while levels below 10 ng/L will lead to the search for adrenal pathology. Values in the “grey zone” are the most challenging since patients with both CD and adrenal pathologies might have intermediate values.
With readily detectable ACTH, then the patient either has CD or an ectopic source. Not infrequently, it is difficult to differentiate between them, as both are ACTH-dependent. The rationale for the tests used is that the corticotroph tumour cells in pituitary adenomas retain some responsiveness to the negative feedback effects of glucocorticoids, while tumours ectopically secreting ACTH generally do not [1, 7, 13, 16]. A rise in cortisol and ACTH to corticotropin-releasing hormone (CRH) test (either alone or in combination with desmopressin) usually suggests CD. An ACTH increase >35 % and cortisol >20 % above baseline levels is considered to be a specific response for CD when ovine-CRH is used [32], and >105 % and >14 %, respectively when human-CRH is used [33]. The CRH test has a sensitivity of 94 % for cortisol and ACTH responses, but approximately 5–17 % of patients with ECS respond to CRH administration [7, 16, 32, 33]. Using desmopressin instead of CRH, 40 % false-positive responses were observed in patients with ECS, with a reported sensitivity of 77–84 % and specificity 73–83 % [7, 16, 34, 35]. The high-dose dexamethasone suppression test (HDDST) is no longer in widespread use, but a serum cortisol suppression greater than 50 % is considered indicative of CD with sensitivity of 60–100 % and a specificity of 65–100 % [16, 18, 35]; when a cut-off for serum cortisol suppression was >60 % this occurred in 3 % of patients with ECS, and when it was >80 % it did not occur in any patient with EAS [16, 18, 35, 36]. The crucial next step to accurately identify a pituitary source is by direct sampling of the effluent of the pituitary by bilateral inferior petrosal sinus sampling (BIPSS) to CRH stimulation; especially as these tumours are usually microadenomas and may not be visible on magnetic resonance imaging (MRI) [1, 16]. This may be considered as the “gold standard” test unless there is a clear pituitary abnormality (macroadenoma), or if an ectopic source has been considered unlikely or the patient is too ill and requires immediate medical therapy. The criteria used to identify CD is an inferior petrosal sinus-to-peripheral ACTH ratio at baseline >2.0 and a gradient >3.0 following CRH (or desmopressin as a cheaper alternative) stimulation [1]. In patients with ECS, both ratios are expected <2.0 but there are reports of false negative and false positive responses, particularly when an adequate hypercortisolaemic state is not present [7, 19, 37–41]. This test demands experience and should be performed only in specialised centres since serious complications (stroke, perforation of the arterial wall, haematoma and transitory arrhythmias) have been described [42]. As previously mentioned, ECS can also have a rapid onset and a paraneoplastic wasting syndrome which may mask hypercortisolism features; profound hypokalemia and less often hyperglycaemia may reveal its presence. Hypokalaemia is related to the degree of hypercortisolaemia since cortisol acts as mineralocorticoid because of the saturation of the enzyme 11 β-hydroxysteroid dehydrogenase type 2. With regard to ACTH and potassium levels, no cut-off limit has been defined to distinguish patients with ECS and CD [7] since cortisol-secreting macroadenomas may share a common biochemical profile [43]. Measurement of circulating tumour markers also has a role when ECS associated with NETs is suspected. Calcitonin and gastrin have been both found to be the most commonly elevated tumour markers, regardless of tumour type [7] in ECS, while calcitonin and catecholamines need to be measured to exclude an ECS source from MTC or a phaeochromocytoma, respectively. Axial imaging with thin-cut multislice-computed tomography (CT) of the chest, abdomen or with chest and pelvis MRI have the highest detection rate for ECS source identification [7, 9, 19, 44]. MRI of the abdomen is not used routinely because of the bowel movement artefact and because calcification associated with the primary tumour can more easily be identified on a CT scan; CT and MRI failure to localise the source of ECS may fall from a maximum reported 50 % to 12.5 % when an appropriate and meticulous technique is used along with a close prolonged follow-up using additional newer imaging modalities when appropriate [7]. Somatostatin receptor (SSTRs) scintigraphy may be proved helpful as ECS may be caused by small NETs expressing SSTRs adding supportive functional data to the conventional imaging techniques [7]. Positron emission tomography (PET) with 18-fluorodeoxyglucose (FDG-PET) may be proved of utility only in very occasional cases since these tumours have usually low metabolic activity [45]. Whole-body PET with 11C-5-hydroxytryptophan has been proposed as universal imaging technique for NETs, in particular identifying an otherwise occult NET [46]. 131I- and 123I-meta-iodobenzylguanidine scans have been also used in NET investigation [7, 9]. All these techniques may also be useful in the investigation of ECS [47]. The aforementioned modalities have resulted in only a limited use for whole-body venous catheterisation and sampling, while selective abdominal angiography and endoscopic ultrasonography may be useful in suspicion of a pancreatic NET [48] and thyroid ultrasound with fine needle aspiration may be used to exclude or diagnose MTC [49]. More sophisticated imaging techniques with limited experience such as the use of an intraoperative gamma counter in the management of metastatic ACTH-secreting bronchial carcinoids look promising [50]. The appearance of the adrenals on CT scanning may also support a diagnosis, such as the possibility of a phaeochromocytoma as the ECS source; the adrenals may have normal size in 7–25 % of patients with ECS, showing moderate hyperplasia in 56 % and marked hyperplasia in 37 % compared to 50 % with either normal or mildly enlarged adrenals in CD; however, macronodular hyperplasia presents similar rates in both clinical settings [7]. Definitive proof of an ACTH-dependent tumour requires complete resolution of the clinical and biochemical features after tumour resection or partial resolution after tumour debulking and/or demonstration of ACTH immunohistochemical staining in the tumour tissue or in metastatic deposits [1, 7].
On the other hand if ACTH is very low or undetectable, then the next step is imaging of the adrenals. High-resolution CT scanning gives the best resolution of adrenal anatomy and it is accurate for masses >1 cm. A mass >5 cm in diameter should be considered to be malignant until proven otherwise [1, 7].
Less commonly, genetic testing for mutations of PRKAR1A test may be needed to confirm Carney’s complex.
Summarising the more recent international consensus statements for the diagnosis and differential diagnosis of CS, published in 2008 [15] recommends that UFC (≥2 values), LDDST, or late night salivary cortisol (≥2 values) to be used as the first line screening tests. Abnormal results should be confirmed by an additional one of these tests. In patients with discordant results, second-line tests should be used as necessary for confirmation or they should be repeated in a second time if a suspicion of cyclicity has been raised. Once the diagnosis of CS is unequivocal, ACTH levels, CRH testing (combined with LDDST or HDDST), together with appropriate imaging, are the most useful non-invasive investigations to determine the aetiology. BIPSS is recommended in cases of ACTH-dependent CS where the clinical, biochemical or radiological results are discordant or equivocal.
Present and Future Therapies (Fig. 9.2)
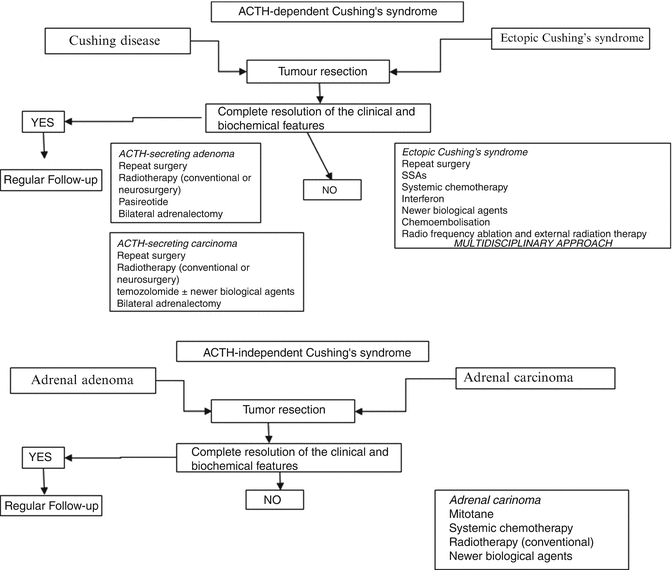
Fig. 9.2
Treatment algorithm in common types of Cushing’s syndrome. ACTH adrenocorticotropin, SSAs somatostatin analogues
Treatment is often complex and may require the use of surgery, radiotherapy and medical management or their combination. The specific target is to normalise cortisol levels with a reversal of clinical features. Surgical removal of the tumour causing CS is the current first-line approach for all types of CS. Inhibitors of cortisol secretion may be required before surgery after the completion of the biochemical diagnostic workup to reverse rapidly the metabolic consequences and poor healing, or in patients who cannot be submitted to surgical procedures, or during the “waiting follow-up period” until the identification of a covert or occult tumour [48], or as alternative treatment if surgery fails [1, 16, 51]. In cases of SCLCs appropriate medical therapy is urgently instituted since prompt correction of hypercortisolism is necessary to minimise the side of myelosuppressive cytotoxic chemotherapy [7]. Compounds that target glucocorticoid synthesis (adrenal secretion inhibitors or adrenolytic drugs, such as aminoglutethimide, metyrapone, ketoconazole, etomidate, mitotane or trilostane) or function (mifepristone) have been broadly used [52]. Metyrapone and ketoconazole (either alone or in combination) are the most frequently used drugs, and appear to be more effective and better tolerated than other adrenal inhibitors. Metyrapone is rapid in onset and highly effective but the presence of hirsutism frequently precludes its use in females; ketoconazole can be used additionally or in place of metyrapone, although its onset of action is rather slower, occurring over several days while gynaecomastia or hypogonadism complicates its use in men. Intravenous etomidate at sub-anaesthetic doses remains an important option when intravenous administration is required for rapid treatment of severely ill patients and is almost always very effective; however, it should usually be used in a intensive care unit in the first instance, while fluconazole use needs to be further evaluated. Finally, if all else fails, the glucocorticoid antagonist mifepristone can reduce the symptoms and signs of CS but serum cortisol cannot be used as marker of efficacy and the patient can become Addisonian unless care is taken [53]; severe hypokalaemia may be induced (treatable with spironolactone) and further evaluation of its safety and follow-up methodologies is needed. Mitotane might be an alternative but the difficulty in monitoring and the adverse effects determine that its use be confined to the minority of patients who are intolerant or not responsive to the previous treatments, and who are unsuitable for adrenalectomy [51]. It is equally important to manage the metabolic problems associated with florid CS. Diabetes needs to be controlled mostly with oral antidiabetics but possibly requiring insulin if severe hyperglycaemia. Present Blood pressure may also be controlled by drug therapy; hypokalaemia, which is seen in almost all patients with ECS and some 10 % of patients with other aetiologies, may be treated by spironolactone or triamterene. The high pro-thrombotic state of patients with CS may be treated by prophylactic doses of heparin. Haloperidol may be used to calm the patient with psychosis and more recently olanzepine has been also used. The hypercortisoloemia patients with ECS are at high risk of sepsis, often with minimal clinical signs, and any such infection bacterial, fungal or viral must be vigorously treated properly as is seen in other immunosuppressed patients along with the management of hypercortisoloemia.
Regarding CD, currently, there is no effective medical therapy that directly and reliably targets the ACTH-secreting pituitary adenoma. Various compounds with neuromodulatory properties have been used to suppress ACTH secretion in patients with CD [51], including gamma-aminobutyric acid (GABA) and serotonin antagonists, but none was proved to be of clinical value. Nuclear hormone receptor ligands involved in hypothalamo–pituitary regulation have been tested; thiazolidinediones were proved to have no effect on pharmaceutical doses used in clinical practice; retinoic acid seems to be of clinical interest since it inhibits ACTH-secretion and cell proliferation both in vitro in ACTH-producing tumour cell lines, and cultured human corticotroph adenomas, and in vivo in nude mice [54]; a very recent clinical trial showed some activity. Only dopamine agonists and somatostatin analogues (SSAs) have shown some promising results. Pasireotide targeting mostly the SSTR5 (but not octreotide and lanreotide targeting the SSTR2) and the dopamine agonist cabergoline both show therapeutic promise in CD. The recent 12-month phase 3 published study of pasireotide showed remission of hypercortisolaemia, as measured by UFC, in 15 % of patients treated by 600 μg twice daily dose, and 26 % of patients on the 900 μg twice daily dose; by increasing the daily dose with an additional 300 μg twice daily, they improved to 16 % and 29 %, respectively [55]. The major side effect was an early rise of blood glucose and glycated haemoglobin followed by stabilisation and the need of a glucose-lowering medication in 46 % of patients [55]. No other medical therapies seem to be reliably effective currently in CD [56]. However, in some ECS cases, SSTRs as well as dopamine receptors have been identified in the primary tumours, and thus their agonists, alone or in combination, have been tried as drug therapy alone or in combination with inhibitors of steroidogenesis in cases of recurrence, incomplete resection or occult tumours [7]. These combinations may also be useful in resistant CD [56].
Selective adenomectomy performed by transsphenoidal surgery (TSS) remains the optimal treatment for ACTH-secreting pituitary adenomas. Remission rates range between 60 % and 80 % with a recurrence rate of 10–25 % after prolonged follow-up [57]; those rates are lower and higher respectively in patients harbouring macroadenomas. Persistent disease might mandate immediate reoperation but this appears to result in remission in only around 50 % of cases, and with a high risk of hypopituitarism [58]. When the tumour is apparent at transsphenoidal exploration, a selective adenomectomy is performed, but when no tumour is obvious a hemi-hypophysectomy as guided by the BIPSS or MRI imaging results is often the best course of action [59]. Postoperative concentration of cortisol <50 nmol/L defines cure but is not predictive of permanent cure; this needs glucocorticoid replacement treatment until recovery of the HPA axis, i.e., when the morning cortisol levels or the cortisol response to an ACTH stimulation test is normal. Higher postoperative cortisol levels are more likely to be associated with failed surgery; however, cortisol levels may sometimes gradually decline over 4–6 weeks reflecting either gradual infarction of remnant tumour or some degree of adrenal semiautonomy [60].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


