Single Photon Emission Computed Tomography (SPECT)
Radionuclide
T 1/2 (h)
Decay mode (%)
E γ (keV) (%)
Production mode
99mTc
6.02
IT (100)
γ
141 (91)
99Mo/99mTc generator
111In
67.9
EC (100)
Auger
γ
171 (90)
245 (94)
Cyclotron
123I
13.2
EC (100)
γ
159 (84)
27 (71)
31 (16)
Cyclotron
Positron Emission Tomography (PET)
Radionuclide
T 1/2 (h)
Decay mode (%)
E β+ (keV) (%)
Production mode
18F
1.83
β+ (97)
EC (3)
634 (97)
Cyclotron
11C
0.34
β+ (99.79)
EC (0.21)
968 (100)
Cyclotron
68Ga
1.13
β+ (89)
EC (11)
1899 (88)
68Ge/68Ga generator
64Cu
12.7
β+ (19)
β− (40)
EC (41)
656 (18)
Cyclotron
This chapter summarizes the established agents used for NET imaging. The main focus is on the imaging of peptide receptors, especially somatostatin receptors, with means of radiolabeled somatostatin analogs, by far the most advanced imaging probes for NETs. This chapter discusses radiopharmaceuticals that have been or are used clinically and refers only selectively to preclinical developments. The chapter is authored from the perspective of the radiopharmaceutical design and development and not from the perspective of their clinical outcome.
7.2 Current Radiopharmaceuticals
7.2.1 Somatostatin Receptor Imaging
Despite the fact that NETs are clinically and pathologically heterogeneous, they share the unique feature of overexpressing somatostatin receptors (sstr). Five somatostatin receptor subtypes are known (sstr1–sstr5) with the sstr2 being the most prominent one in differentiated NETs [4]. However, different subtypes may be expressed concomitantly and in various combinations and frequencies on pathological targets [5, 6]. The somatostatin receptors belong to the family of G-protein-coupled receptors, located on the plasma membrane of the targeted tumor cell. The natural ligand somatostatin consists of two cyclic disulfide-containing peptide hormones, one with 14 amino acids (SS-14, Table 7.2) and one with 28 amino acids (SS-28).
Table 7.2
Somatostatin analogs that have been used in humans
Code | Chemical structure |
|---|---|
SS-14 | Ala-Gly-cyclo(Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys) |
Somatostatin receptor agonists | |
OC | D-Phe-cyclo(Cys-Phe-D-Trp-Lys-Thr-Cys)Thr(ol) |
TOC | D-Phe-cyclo(Cys-Tyr-D-Trp-Lys-Thr-Cys)Thr(ol) |
TATE | D-Phe-cyclo(Cys-Tyr-D-Trp-Lys-Thr-Cys)Thr |
HA-TATE | D-Phe-cyclo(Cys-3-Iodo-Tyr-D-Trp-Lys-Thr-Cys)Thr |
Lanreotide | D-β-Nal-cyclo(Cys-Tyr-D-Trp-Lys-Val-Cys)-Thr-NH2 |
NOC | D-Phe-cyclo(Cys-1-Nal-D-Trp-Lys-Thr-Cys)Thr(ol) |
Somatostatin receptor antagonists | |
BASS | p-NO2-Phe-cyclo(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)D-Tyr-NH2 |
JR11 | Cpa-cyclo[D-Cys-Aph(Hor)-D-Aph(Cbm)-Lys-Thr-Cys]-D-Tyr-NH2 |
Somatostatin itself turned out to be unsuitable for in vivo use as it has a short plasma half-life of about 3 min. Somatostatin analogs more resistant to enzymatic degradation than somatostatin itself were developed by various modifications of the natural molecule. The introduction of d-amino acids for improved in vivo stability, the use of the minimal chain length for conserving the biological activity of the natural hormone, the hexapeptide lead structure Cys-Phe-d-Trp-Lys-Thr-Cys, and the systematic elongation of the N and C terminals allowed the Sandoz team in 1980 to characterize the most stable and active analog known as octreotide (OC) (Table 7.2).
7.2.1.1 Somatostatin Receptor Scintigraphy and SPECT/CT
Octreotide was conjugated at the N-terminus with the acyclic chelator diethylenetriaminepentaacetic acid (DTPA, Fig. 7.1) that is able to coordinate metals such as In3+. Since the mid-1990s, the radiolabeled analog [111In-DTPA0]octreotide (OctreoScan®, Covidien, Petten, The Netherlands) is commercially available for somatostatin receptor scintigraphy. OctreoScan was the first peptide-based nuclear imaging probe approved by the US Food and Drug Administration (FDA) in 1994. The procedure guidelines for tumor imaging with OctreoScan have been described by the European Association of Nuclear Medicine (EANM) [7]. OctreoScan is used mainly in midgut carcinoid, pancreatic NET, and neuroblastoma but also in growth hormone-secreting pituitary tumors, paraganglioma, medullary thyroid carcinoma, pheochromocytoma, meningioma, and Merkel cell tumors [8] and became an integral part of the management of NET patients [9].
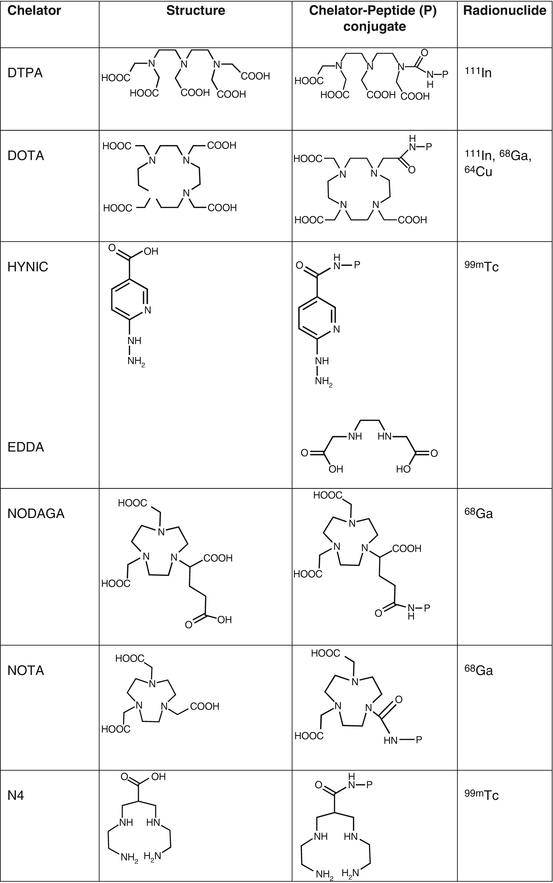
Fig. 7.1
Chelators of clinically used radiopharmaceuticals for neuroendocrine tumors imaging
[111In-DTPA0]octreotide has moderate binding affinity to sstr2 (Table 7.3). The replacement of Phe3 in octreotide by Tyr3 (TOC, Table 7.2) led to an improved sstr2 affinity (Table 7.3), while the C-terminal introduction of Thr8 (TATE, Table 7.2) for Thr(ol)8 (TOC) resulted in a further improvement of the sstr2 affinity (Table 7.3) [10]. On the other hand, DTPA is not a suitable chelator for other 3+ radiometals, such as 68Ga, 90Y, or 177Lu. The chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, Fig. 7.1) has replaced DTPA, as it forms thermodynamically and kinetically stable complexes with a series of 3+ radiometals, including 111In, 68Ga, 90Y, and 177Lu. Both somatostatin conjugates DOTA-TOC and DOTA-TATE, labeled with 111In, appeared to be nearly equivalent in a head-to-head comparison [11]. However, their use in SPECT imaging of NETs has been limited. To circumvent the drawbacks of 111In, such as cost, limited availability, and high radiation burden, TOC was conjugated to an efficient bifunctional chelator for the generator-produced 99mTc, namely, hydrazinonicotinamide (HYNIC, Fig. 7.1). 99mTc-EDDA/HYNIC-TOC (EDDA, ethylenediamine-N,N’-diacetic acid acting as a coligand for 99mTc labeling, Fig. 7.1) was granted marketing authorization and became commercially available in some European countries, under the commercial name 99mTc-Tektrotyd. Later on, 99mTc-EDDA/HYNIC-TATE was also developed. The two 99mTc-labeled analogs have similar performance and seem to have better or at least comparable sensitivities to OctreoScan [12–15].
Table 7.3
Affinity profiles (IC50) for human sstr1–sstr5 of a series of somatostatin analogs and their metallated conjugates that have been used clinically in neuroendocrine tumors imaging
Somatostatin analog | sstr1 | sstr2 | sstr3 | sstr4 | sstr5 |
|---|---|---|---|---|---|
Agonists | |||||
DTPA-octreotidea | >10,000 | 12 ± 2 | 376 ± 84 | >1000 | 299 ± 50 |
In-DTPA-octreotidea | >10,000 | 22 ± 3.6 | 182 ± 13 | >1000 | 237 ± 52 |
DOTA-TOCa | >10,000 | 14 ± 2.6 | 880 ± 324 | >1000 | 393 ± 84 |
Ga-DOTA-TOCa | >10,000 | 2.5 ± 0.5 | 613 ± 140 | >1000 | 73 ± 21 |
DOTA-TATEa | >10,000 | 1.5 ± 0.4 | >1000 | 453 ± 176 | 547 ± 160 |
Ga-DOTA-TATEa | >10,000 | 0.20 ± 0.04 | >1000 | 300 ± 140 | 377 ± 18 |
DOTA-lanreotidea | >10,000 | 26 ± 3.4 | 771 ± 229 | >10,000 | 73 ± 12 |
Ga-DOTA-NOCb | >10,000 | 1.9 ± 0.4 | 40.0 ± 5.8 | 260 ± 74 | 7.2 ± 1.6 |
Antagonists | |||||
DOTA-BASSc | >1000 | 1.5 ± 0.4 | >1000 | 287 ± 27 | >1000 |
In-DOTA-BASSc | >1000 | 9.4 ± 0.4 | >1000 | 380 ± 57 | >1000 |
DOTA-JR11d | >1000 | 0.72 ± 0.12 | >1000 | >1000 | >1000 |
In-DOTA-JR11d | >1000 | 3.8 ± 0.7 | >1000 | >1000 | >1000 |
Ga-DOTA-JR11d | >1000 | 29 ± 2.7 | >1000 | >1000 | >1000 |
Lu-DOTA-JR11d | >1000 | 0.73 ± 0.15 | >1000 | >1000 | >1000 |
NODAGA-JR11d | >1000 | 4.1 ± 0.2 | >1000 | >1000 | >1000 |
Ga-NODAGA-JR11d | >1000 | 1.2 ± 0.2 | >1000 | >1000 | >1000 |
Historically, the first 99mTc-based somatostatin analog was 99mTc-depreotide (NeoTect (Diatide Inc.); NeoSpect (Nycomed)), with the peptide sequence Lys-Cys-Lys-(beta-DAP)-CH2CO-S-cyclo(hCys-(N-Me)Phe-Tyr-Trp-Lys-Val) that is actually a non-octreotide-based analog. FDA approved 99mTc-depreotide in 1999 for the detection of lung cancer in patients with pulmonary nodules [16]. However, its use was rather constrained, compared to OctreoScan, mainly due to the relatively high activity in the abdomen limiting its value in the diagnosis of abdominal NETs. 111In-DOTA-lanreotide (Table 7.2) is another somatostatin receptor imaging agent with a slight different affinity profile than OctreoScan (Table 7.3) [10]. In comparison with OctreoScan, it has a lower sensitivity in demonstrating neuroendocrine tumors, but it may have advantages in other tumors, for instance, in differentiated thyroid cancer [17]. 99mTc-depreotide and 111In-DOTA-lanreotide are not ideally suited for imaging abdominal NET primarily because of their diminished sensitivity compared to OctreoScan. OctreoScan has covered for many years the medical need of imaging NETs, and many clinical centers still use it, not only because it is readily available but also because it may adequately answer the question whether a patient is suitable for radionuclide therapy. However, somatostatin receptor imaging is rapidly “switching” to the use of PET tracers.
7.2.1.2 Somatostatin Receptor PET/CT
Within the last 10 years, the fast progression in NET imaging came together with the accelerated development of 68Ga radiochemistry/radiopharmacy. It combined, on one side, the advantages of PET over SPECT (100–1000-fold higher sensitivity, absolute quantitation, and shorter scanning time) and, on the other side, the advantages of using 68Ga-based radiopharmaceuticals [18–21]. 68Ga is produced from a long-lived 68Ge/68Ga generator that makes it available, rather inexpensive and independent of an on-site cyclotron or a reactor. Ultimately, 68Ga radiopharmacy is possible in each hospital. 68Ga has a short half-life of 68 min that is compatible with the relatively fast uptake mechanism of small peptides, such as the somatostatin analogs, that are transported to target tissues via the blood circulation. Thus, compared to 111In, it allows investigations to be completed within 1–3 h while decreasing the radiation dose to the patient by a factor 2–3. Impressive progress in 68Ga radiochemistry and chelation chemistry has been achieved over the last few years due to the success and high potency of the first 68Ga radiopharmaceuticals, especially the first 68Ga-labeled somatostatin analog DOTA-TOC [22, 23]. Logically, 68Ga-labeling of DOTA-TATE followed. Between these two radiopharmaceuticals, 68Ga-DOTA-TATE showed a tenfold higher affinity to sstr2 in vitro, compared with 68Ga-DOTA-TOC (Table 7.3) [10]. Despite its higher affinity, 68Ga-DOTA-TATE was not proven to be superior to 68Ga-DOTA-TOC in PET/CT imaging of NET patients, on the contrary, an opposite tendency was observed [24, 25]. Very recently, the so-called high-affinity TATE (HA-TATE, a TATE analog in which Tyr3 is iodinated, Table 7.2) labeled with 68Ga has been proposed as an adequate, freely available substitute for 68Ga-DOTA-TATE since its use is not governed by patent restrictions [26]. The sstr2 affinities of Ga-HA-DOTA-TATE and Ga-DOTA-TATE (IC50 = 1.4 ± 0.8 nM and 1.2 ± 0.6 nM, respectively) were nearly identical [27].
Modifications of the octreotide at position 3 by introducing the unnatural amino acid 1-naphtyl-alanine (1-NaI) led to the analog NOC (Table 7.2), with affinity not only for the sstr2 but also for sstr3 and sstr5 (Table 7.3) [28, 29]. In theory, 68Ga-DOTA-NOC may have the advantage over 68Ga-DOTA-TOC and 68Ga-DOTA-TATE by targeting a broader spectrum of various tumors or by increasing the tumor uptake in tumor cell coexpressing simultaneously several receptor subtypes. However, head-to-head comparative studies of 68Ga-DOTA-NOC vs 68Ga-DOTA-TATE have shown discrepant results [30, 31].
All three somatostatin conjugates DOTA-TOC, DOTA-TATE, and DOTA-NOC, labeled with 68Ga, are routinely used for PET imaging of NET patients in many hospitals, especially in Europe [32]. The European Association of Nuclear Medicine (EANM) has currently published procedure guidelines on PET/CT tumor imaging using any of the three mentioned PET tracers [33]. Currently, there is no strong clinical evidence on the superiority of one analog over the others [34]. None of the three PET tracers is registered, and both, 68Ga-DOTA-TOC and 68Ga-DOTA-TATE, have received “Orphan Drug Designation” as diagnostic agents for the management of patients with neuroendocrine tumors (NETs) by the US Food and Drug Administration. It also exists a European Pharmacopeia monograph for “gallium (68Ga) edotreotide injection” (No 2482) and one for “gallium (68Ga) chloride solution for radiolabeling” (No 2464).
Recently, DOTA-TATE labeled with 64Cu has been used for PET imaging of NETs [35, 36]. The rational of using 64Cu, instead of 68Ga, lies on delayed imaging due to longer half-life of 64Cu (12.7 h, see also Table 7.1), better resolution, and avoiding the in-house preparation shortly before application. 64Cu-DOTA-TATE was shown to be by far superior to OctreoScan in the diagnostic performance in NET patients in a head-to-head comparison [35]. Whether 64Cu-labeled analogs have any advantage over the 68Ga-labeled counterparts is not known since so far not head-to-head comparison has been performed. However, one could argue that DOTA is far from being an ideal chelator for 64Cu, due to the possible transchelation of 64Cu and therefore the rather limited in vivo stability of the 64Cu-DOTA complex [37, 38]. Indication of the in vivo instability is the accumulation of radioactivity in the liver over time, as shown in many preclinical studies. That may explain the moderate to high liver uptake initially observed in patients where 64Cu-DOTA-TATE was administered [36]. Nowadays, a number of chelators tailored for 64Cu are available, and the in vivo stability of 64Cu-labeled somatostatin analogs can be significantly improved, important especially when delayed images are desired. On the other hand, one should be aware that the choice of the chelator, but also of the radiometal, may influence critical properties of the radiotracer, such as affinity, pharmacokinetics, and others [10, 29, 39, 40].
There is no doubt that 68Ga-labeled somatostatin analogs represent a significant evolution in somatostatin receptor imaging over OctreoScan. They have shown higher sensitivity compared with OctreoScan [41] but also with other tracers used for imaging of NETs that will be discussed later, such as 18F-FDG (particularly in well-differentiated tumors) [42, 43], 18F-DOPA [44–46], and also 123I-MIBG [47, 48]. It seems reasonable that, if available, PET/CT with 68Ga-labeled somatostatin analogs should be the image modality of choice for somatostatin receptor imaging in the future. Moreover, the preparation of 68Ga radiopharmaceuticals is amenable to automation and approaches the kit-type preparation.
The success of 68Ga-DOTA-somatostatin analogs also relies on the fact that the chelator DOTA forms stable complexes with important therapeutic radionuclides, such as 177Lu, 90Y, and others. This allows the same DOTA-conjugated analog to be used either for PET imaging, when labeled with 68Ga, or for Peptide Receptor Radionuclide Therapy (PRRT), when labeled with a therapeutic radionuclide. This theranostic approach, in analogy to what has been used for decades with the various isotopes of iodine in the field of thyroid cancer, has been extremely valuable in the field of NET especially in the era of precision medicine. Somatostatin receptor imaging has paved the way for other radionuclide imaging systems and particularly in the development of different peptide-based radiopharmaceuticals (see Sects. 7.3.2 and 7.3.3).
7.2.2 Metabolic Imaging
7.2.2.1 18F-Fluorodeoxyglucose (18F-FDG) PET/CT
The most widely used PET radiopharmaceutical in daily practice is 2-deoxy-2-(18F)fluoro-d-glucose or 18F-fluorodeoxyglucose (18F-FDG), a glucose analog in which the 2’ hydroxyl group (−OH) has been substituted by 18F (Fig. 7.2). 18F-FDG is taken up by glucose transporters and phosphorylated by the hexokinase enzyme. Once phosphorylated it is not further metabolized thus trapped in the cell [15]. 18F-FDG was actually the first PET radiopharmaceutical to be used for detecting NETs [49]. However, in contrast to the wide use of 18F-FDG in numerous types of tumors, its role in imaging of NETs is rather limited. The well-differentiated NETs (i.e., most GEP-NETs) have a normal or slightly increased glucose metabolism, and the sensitivity of 18F-FDG is rather low for their detection. Therefore, 18F-FDG is not routinely used for this purpose. In undifferentiated or poorly differentiated NETs that exhibit aggressive histologic features, 18F-FDG PET seems to be the best choice since the expression of somatostatin receptors is less frequent, and therefore somatostatin receptor imaging is often negative [42, 43, 50]. Actually, 18F-FDG PET imaging seems to have a complementary role with the first-choice diagnostic tool that is PET imaging with 68Ga-labeled somatostatin analogs [42, 43, 51]. Negative 18F-FDG PET results are predictive for low aggressiveness and a high survival rate, and conversely positive 18F-FDG PET results identified cases with worse outcome [52–54]. Therefore, 18F-FDG may retain an important role in managing NET patients mainly due to its prognostic value and its higher sensitivity in aggressive and high-grade tumors, rather than its role in the diagnosis and staging of NETs.
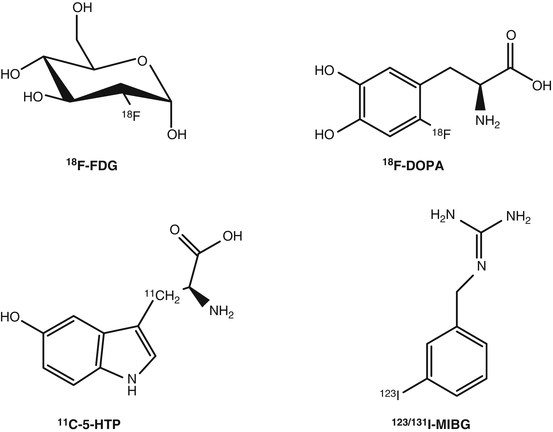
Fig. 7.2
Radiopharmaceuticals used for metabolic imaging of neuroendocrine tumors
7.2.2.2 18F-L-Dihydroxyphenylalanine (18F-DOPA) PET/CT
18F-l-dihydroxyphenylalanine (18F-DOPA) (Fig. 7.2) was initially introduced for studying the physiology and physiopathology of dihydroxyphenylalanine (DOPA) biodistribution in the human brain, in particular Parkinson’s syndrome and then in oncology for imaging NETs or brain tumors. The use of 18F-DOPA in NETs is based on the fact that neuroendocrine tumor cells take up and transport into the cytoplasm the dopamine precursor 18F-DOPA via the cell membrane-bound l-type amino acid transporter (LAT) that is upregulated and highly active in these cells. Inside the cell 18F-DOPA is decarboxylated (metabolized) by the aromatic acid decarboxylase (AADC) to 18F-dopamine, which is transported into storage granules by vesicular monoamine transporter and trapped intracellularly [55]. 18F-DOPA PET has shown very high sensitivity for both staging and restaging of NETs [56]. Compared to somatostatin receptor scintigraphy, 18F-DOPA performs superior for the imaging of NETs, especially in carcinoid patients [57, 58]. Compared to PET imaging using 68Ga-labeled somatostatin analogs, 18F-DOPA performed inferior in NET patients [44–46]. 18F-DOPA PET showed relatively high sensitivity in congenital hyperinsulinism [59] and can be useful to study benign insulinomas, paragangliomas, and pheochromocytomas due to the variable expression of sstr in these tumors. Furthermore, 18F-DOPA can be an option for imaging of serotonin-secreting, sstr-negative NETs.
The production of 18F-DOPA by electrophilic fluorination requires modest radiochemistry expertise and is not widely established. However, nowadays, more and more centers are capable of producing 18F-DOPA, and it is commercially available in several European countries. Contrary to 68Ga-labeled somatostatin analogs, 18F-DOPA lacks of a therapeutic counterpart.
7.2.2.3 11C-5-HTP
The PET radiopharmaceutical β-[11C]-5-hydroxyl-l-tryptophan (11C-5-HTP) (Fig. 7.2) is taken up via the l-amino acid transporter (LAT), it is decarboxylated by the aromatic l-amino acid decarboxylase ADCC, and the end product, serotonin, is transported via the vesicular monoamine transporter (VMAT) and stored into secretory vesicles. 11C-5-HTP visualizes the serotonin pathway, which is active in many NETs. The published clinical results, even though limited, justify its use [57, 60–63]. In comparison with 18F-DOPA and OctreoScan, 11C-5-HTP seems to be best in pancreatic islet cell tumors, while 18F-DOPA in staging of carcinoids [57], and it outperforms OctreoScan in the lesion-based detection, with high sensitivity in small NET lesions, such as primary tumor [63].
11C-5-HTP-PET/CT is a useful imaging technique, with higher sensitivity for pancreatic NETs, as opposed to carcinoids; however its routine clinical use is debatable. The synthesis of 11C-HTP is very complex and it is only produced in a few specialized centers worldwide. Additionally, the short half-life of 11C (t1/2 = 20 min, see also Table 7.1) demands an on-site cyclotron and production in close relation to the scan. These factors severely restrict its clinical use. Similarly to 18F-DOPA, 11C-5-HTP lacks of a therapeutic counterpart.
Related posts:
 Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
Molecular Genetics of Gastroenteropancreatic Neuroendocrine Tumours
 Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors
Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors
 Molecular Genetics of MEN1-Related Neuroendocrine Tumors
Molecular Genetics of MEN1-Related Neuroendocrine Tumors
 Nanoparticles for Radionuclide Imaging and Therapy: Principles
Nanoparticles for Radionuclide Imaging and Therapy: Principles
 Radionuclide Imaging of Medullary Thyroid Carcinoma
Radionuclide Imaging of Medullary Thyroid Carcinoma
 Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Radionuclide Imaging of Gastrointestinal Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


