35 Craniopharyngiomas
Craniopharyngiomas are benign epithelial neoplasms of the sellar region that arise from embryonic squamous cells of the hypophysiopharyngeal duct (Rathke’s pouch). Although Erdheim originally described these lesions in 1904, Cushing introduced the term craniopharyngioma in 1932 to describe these epithelial neoplasms and to denote their origin from the embryological remnant. These tumors are characterized by slow growth that may involve important neurovascular structures. Clinical presentation may be associated with endocrine, visual, and mental disturbances; these are secondary to involvement of the hypothalamus–pituitary axis, optic pathways, thalamus, and frontal lobes. Although advances in microsurgical and skull base techniques, radiotherapy, chemotherapy, and hormonal replacement have provided better long-term survival and longer recurrence-free intervals, controversies remain as to the optimal treatment of these tumors.
Typically, craniopharyngiomas are located in the parasellar region. Approximately 5 to 15% manifest within the confines of the sella.1,2 Another 20% present as a suprasellar mass.2 Their pattern of growth and location largely depend on where they arise from the pituitary stalk. Lesions arising from the distal portion of the stalk may grow within the sella turcica. They usually adhere to a midline location and may extend into the suprasellar compartment and third ventricle. Lesions arising from the more proximal end of the stalk may grow predominantly within the third ventricle.3 Thirty percent of craniopharyngiomas may extend anteriorly to involve the frontal lobes; 25% grow laterally to involve the temporal lobe and structures of the middle cranial fossa; another 20% may grow posterior and inferior to encroach on the brainstem and may extend into the cerebellopontine angle or foramen magnum.2 Papillary craniopharyngiomas more often are located in the third ventricle.4
 Incidence
Incidence
Craniopharyngiomas have an annual incidence of 0.5 to 2.0 cases/million population per year and are seen in both adults and children.5,6 These tumors account for 1.2 to 4% of adult intracranial tumors and 6 to 10% of intracranial neoplasms in the pediatric population.7,8
Craniopharyngiomas exhibit a bimodal age distribution, with the first peak at age 5 to 10 years and second peak between ages 50 and 60 years. They occur equally between sexes.7 The classic adamantinomatous subtype occurs 10 times as often as the papillary subtype and mainly in children.4 In contrast, the papillary subtype only occurs in adults.4
 Embryology: Derivation from Rathke’s Pouch
Embryology: Derivation from Rathke’s Pouch
At approximately the fourth week of gestation, Rathke’s pouch forms as a diverticulum of the embryonic stomodeum (roof of the oral cavity). Rathke’s pouch migrates upward to meet with the infundibulum, which is a down-growth from the floor of the diencephalon. The migratory path of Rathke’s pouch corresponds with the primitive craniopharyngeal duct. At approximately the second month of fetal life, Rathke’s pouch separates to form Rathke’s vesicle, which surrounds the infundibulum. The cells that compose Rathke’s vesicle eventually form the pars distalis, pars tuberalis, and pars intermedia, which compose the adenohypophysis. Craniopharyngiomas were originally believed to originate from squamous cell rests found along the path of the primitive craniopharyngeal duct and adenohypophysis at the surface of the pituitary stalk,4 but now it is thought that those cells are not remnants of the craniopharyngeal duct, but actually result from metaplasia of adenohypophyseal cells of the pituitary stalk.9 These cells have an increased frequency with age and thus cannot be responsible for the largely juvenile onset of craniopharyngiomas.4
Craniopharyngiomas of both subtypes are thought to derive from Rathke’s cleft/pouch as evidenced by the occasional ability of some tumor cells to express one or more pituitary hormones.4 It has also been suggested that the adamantinomatous craniopharyngioma may arise from embryonic rests with enamel organ potential.4,10 Papillary craniopharyngiomas may share a similar origin or represent a spectrum of a similar disease process with Rathke’s cleft cysts as evidenced by the fact that these two disease entities are occasionally of very similar pathology.4 Papillary craniopharyngiomas may have focal ciliation of epithelium or goblet cells, whereas some Rathke’s cleft cysts have extensive squamous metaplasia of their cyst wall, resulting in a solid component, and these cysts may have an associated higher rate of recurrence more similar to papillary craniopharyngiomas.4,10
Special Consideration
• Craniopharyngiomas are no longer thought to arise from squamous cell rests found along the path of the primitive craniopharyngeal duct and adenohypophysis but are derived from Rathke’s cleft.
• There is debate as to whether the two pathological variants of craniopharyngioma, adamantinomatous and squamous papillary type, are of common or separate origin. Some tumors demonstrate the presence of both adamantinomatous and squamous papillary elements.
 Genetics
Genetics
Typically, craniopharyngiomas occur sporadically and do not follow a direct pattern of familial inheritance; rare cases of craniopharyngioma occurring in siblings, cousins, and children of an affected parent have been reported. Studies have shown that a subset of adamantinomatous craniopharyngiomas are of a monoclonal origin and, thus, are due to somatic genetic defects at specific chromosomal loci.11 No consistent genetic abnormalities have been observed, and the precise molecular mechanisms involved in craniopharyngioma development are unknown11; however, several genetic abnormalities have been reported. Beta-catenin gene mutations have been described in adamantinomatous craniopharyngiomas but not in the papillary type.12,13 All adamantinomatous craniopharyngiomas demonstrate expression of beta-catenin and some have beta-catenin mutations, which activate the Wnt signaling pathway, causing mitogenic stimulation and mis-specification of cells to form distinct structures, including increased expression of enamel proteins.12,13 Adamantinomatous craniopharyngiomas have shown other gene changes. One study found that six of nine tumors displayed at least one genomic alteration and three had six or more alterations; the most common abnormality was chromosomal gains.11 Loss of the Y chromosome has also been observed in some craniopharyngiomas. In contrast, no tumors demonstrated chromosomal imbalances or changes in DNA copy number in 20 adamantinomatous and nine papillary craniopharyngiomas evaluated in another study.14
 Pathology
Pathology
Adamantinomatous Type
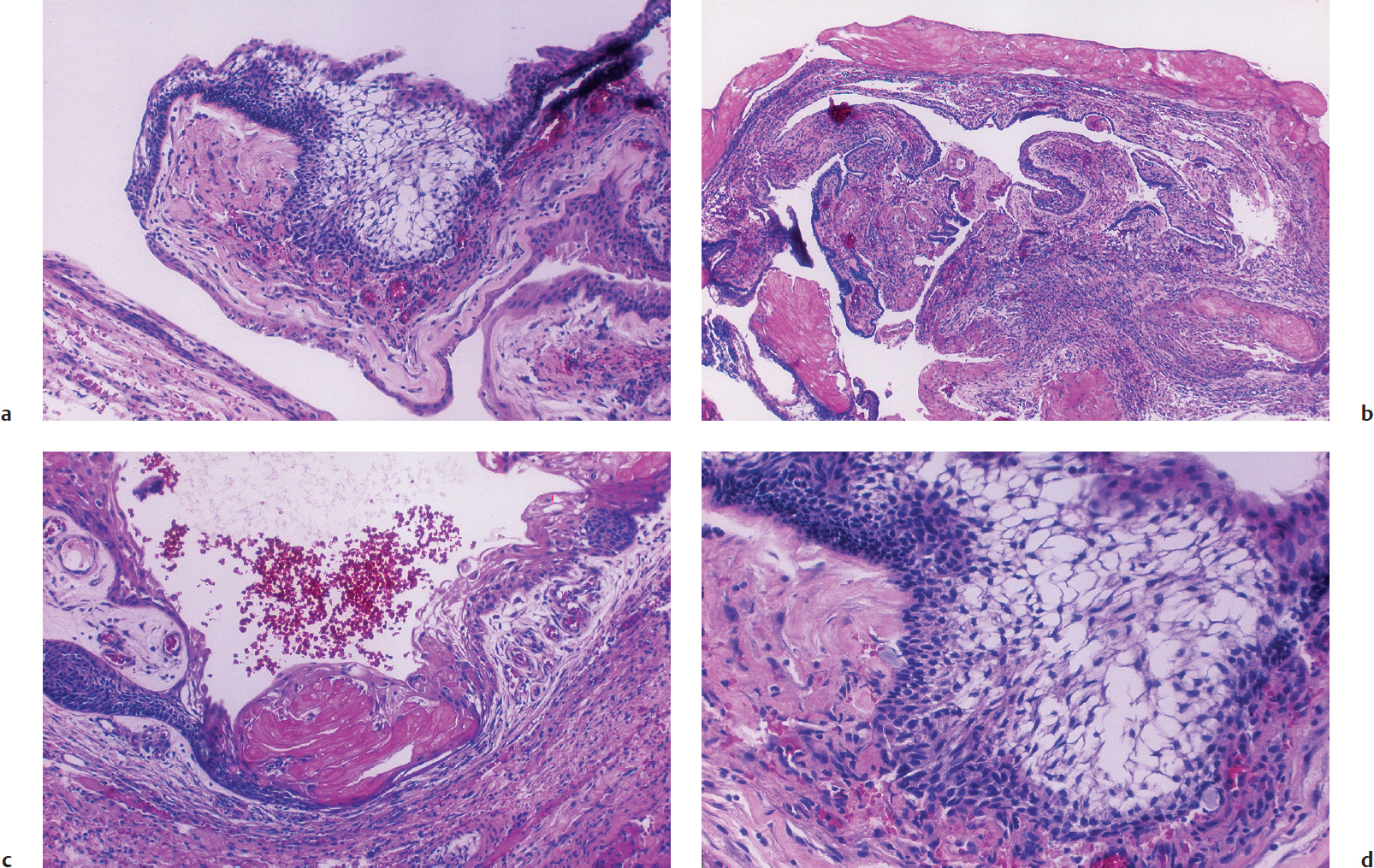
The adamantinomatous or “childhood” type of craniopharyngioma resembles a neoplasm of tooth-forming tissues10 and is grossly cystic with a smaller solid component with calcification. The cystic fluid contains cholesterol and necrotic debris that impart a dark brown to black “motor oil” appearance to the fluid. The cystic fluid is believed to be secondary to desquamation of the cyst epithelium. A well-described pathological feature of the adamantinomatous type is its propensity to adhere to blood vessels and adjacent neural structures, especially the hypothalamus.4 It commonly has a ragged interface with the surrounding anatomy.4 This adherence promotes a glial reaction at the tissue–tumor interface characterized by an intensive gliosis with Rosenthal fiber formation.4 This localized process provides a plane for dissection during surgical removal.
Microscopically, the epithelium is arranged in a distinctive adamantinomatous-like pattern. This pattern is characterized by a basal layer containing cells with darkly staining nuclei associated with an intermediate layer of stellate cells surrounded by a layer of columnar epithelium (Fig. 35.1). Histological examination of the cyst wall reveals a keratinized squamous epithelium with discrete areas of stacked clusters of desquamated cells, giving the appearance of nodules of keratin (“wet keratin”). It is the mineralization and deposition of calcium salts to this keratin-rich epithelium that accounts for the calcification seen in this neoplasm.
Papillary Type
In contrast to the adamantinomatous variety, which may occur in children and adults, the squamous papillary type occurs almost exclusively in adulthood, with only rare isolated cases reported in children. It has a distinct propensity to involve the third ventricle.4 Upon gross examination, the squamous papillary type is mostly solid but may possess a cystic component.4 Microscopically, the squamous papillary variant exhibits nests of well-differentiated keratinized stratified squamous epithelium forming papillae. These tumors are well circumscribed, rarely calcify, and lack the “wet keratin” and motor oil content seen in the adamantinomatous form.4 In addition, they do not adhere to surrounding structures; however, gliosis and Rosenthal fiber formation may be present. Unlike the epithelium found in the classic craniopharyngioma, the papillary form does not resemble the tissues of enamel organs.
 Molecular Markers
Molecular Markers
The identification of molecular markers for craniopharyngiomas has begun, but their clinical use has not been identified. A subset of craniopharyngiomas have increased insulin-like growth factor I receptor (IGF-IR) expression and display growth arrest with IGF-IR inhibitors.15 In craniopharyngiomas that express estrogen and progesterone receptors (~30%), the incidence of regrowth after surgery is higher in patients negative for these receptors because of loss of differentiation.16 An elevated β-human chorionic gonadotrophin level has been reported in the cerebrospinal fluid (CSF) of patients with craniopharyngioma, and the tumor has been found to stain positive for this hormone. Finally, a high MIB-1 staining (Ki-67 index) suggests a high possibility of tumor regrowth and was significantly higher in patients who progressed to have a recurrence compared with patients without regrowth.16
 Clinical Features
Clinical Features
Presentation
Clinical manifestations of craniopharyngiomas depend on the origin, direction of growth, degree of tumor extension, and involvement of surrounding neural structures. Patients may present with symptoms related to increased intracranial pressure from mass effect or hydrocephalus, or from compression of the optic apparatus, pituitary-hypothalamus axis, or cerebrum. Headache is encountered in more than 50% of patients and is one of the most common complaints prompting medical attention in all age groups. Children often tolerate a significant degree of visual loss; it is the headaches, vomiting, and behavioral changes that usually bring them to initial medical attention. Adults more often than children present with visual disturbance, usually in a delayed fashion because of macular sparing and because they may erroneously relate it to external causes.17
Pearl
• Patients with craniopharyngioma typically present with symptoms related to increased intracranial pressure from mass effect or hydrocephalus, or from compression of the optic apparatus, pituitary-hypothalamus axis, or cerebrum. Headache is one of the most common complaints prompting medical attention in all age groups.
Visual Symptoms
Although approximately 20% of children have papilledema at presentation,1 adults appear to be more sensitive to visual deficits than children. Eighty percent of adults demonstrate visual disturbance as a presenting symptom.1 Visual disturbance may manifest as a decrease in visual acuity,5 diplopia, blurred vision, bitemporal hemianopia, homonymous hemianopia, various quadrantanopsias, or seesaw nystagmus.17 Rare cases of unilateral or even bilateral blindness have been reported.
Endocrine Abnormalities
Endocrine abnormality is found in 80 to 90% of individuals at presentation.1,7,18 Children commonly present with short stature and delayed linear growth, whereas adolescents may present with delayed or arrested puberty. Men may observe a loss of libido; women may present with secondary amenorrhea.
The most common hormonal deficiencies include growth hormone (75%), followed by luteinizing hormone or follicle-stimulating hormone (40%), adrenocorticotropic hormone (25%), and thyroid-stimulating hormone (25%). The presence of hyperprolactinemia in 20% of patients indicates impingement of areas within the hypothalamus or the pituitary stalk that normally exert an inhibitory influence on prolactin release (“stalk section” effect). Hypothalamic compression may occasionally lead to precocious puberty owing to loss of hypothalamic inhibition exerted on the gonadotropin-releasing hormones in the preadolescent phase. Precocious puberty, however, may be offset by panhypopituitarism secondary to compression of the pituitary gland. Diabetes insipidus (DI) is infrequent at presentation, occurring in 9 to 17% of patients before surgery.18
Pearl
• Diabetes insipidus is a common feature in patients postoperatively; however, fewer than 20% of patients manifest DI at presentation.
Behavioral Changes
Some patients come to medical attention as a result of changes in mental status or behavior. Although unusual in children, about 25% of adults present with mental disturbance.17
Psychological or intellectual manifestations are largely due to the direction of tumor expansion. Tumor growth involving the frontal lobes may cause dementia, apathy, abulia, or psychomotor slowing.1 Three of 12 patients presenting with mental status change demonstrated characteristics of Korsakoff syndrome in one report.19 Complex psychomotor seizures and amnesia have been documented with tumor extension into the temporal lobe and hippocampus.1
Imaging Studies
Plain radiographs of the skull have largely been replaced by computed tomography (CT) or magnetic resonance imaging (MRI) as the initial imaging study for diagnosis of craniopharyngioma. Approximately 66% of adults and more than 90% of children, however, exhibit some abnormality on plain skull X-ray, such as enlargement of the sella, erosion of the clinoids and dorsum sella, or suprasellar calcification. More than 80% of children and 40% of adults show calcification on plain skull radiographs.1
Computed tomography demonstrates calcification (Fig. 35.2) and the secondary skull base bone changes.20 Calcifications are seen in 93% of childhood craniopharyngiomas.20 The cyst fluid is iso- or hypodense on CT but may appear hyper-dense if sufficient calcification is present. CT with intravenous contrast results in enhancement of the solid portion of the tumor as well as the cyst capsule.
Magnetic resonance imaging is the neuroimaging modality of choice, precisely demonstrating the extent and location of the tumor as well as the tumor’s relationship to important surrounding neurovascular structures. Cystic components (Fig. 35.3) are identified in 54 to 94% of all craniopharyngiomas,21 but they are found in 99% of pediatric craniopharyngiomas.20 On MRI, the cyst exhibits a hyperintense signal on T1-weighted images. The solid component is isointense but enhances on administration of intravenous gadolinium. Magnetic resonance (MR) or CT angiography provides anatomic detail of the cerebral vasculature in relation to the tumor, which is important in surgical planning.
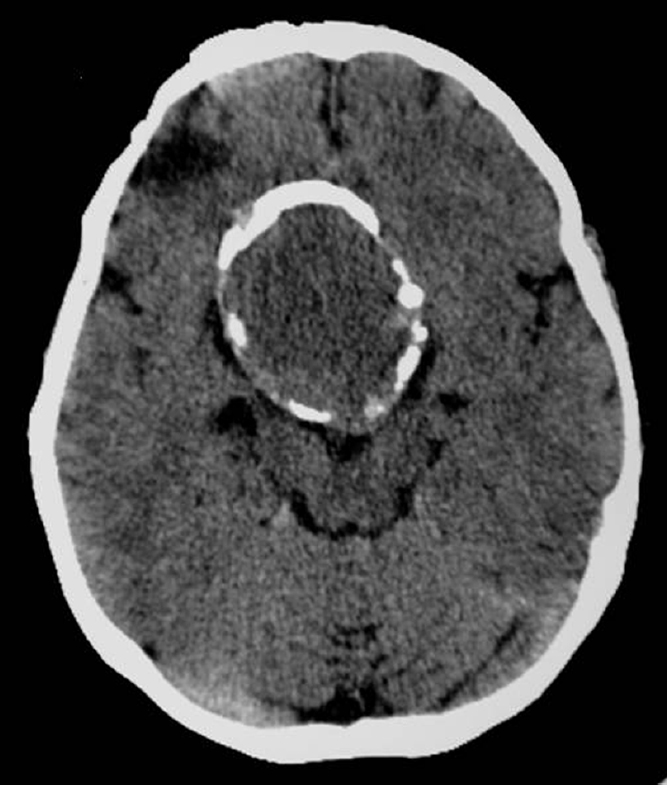
Fig. 35.2 Imaging of a 48-year-old patient who had a subtotal resection of a craniopharyngioma at age 5 and 43 years later experienced increasing headaches and decline in vision. Computed tomography demonstrated a suprasellar lesion with a calcified rim.
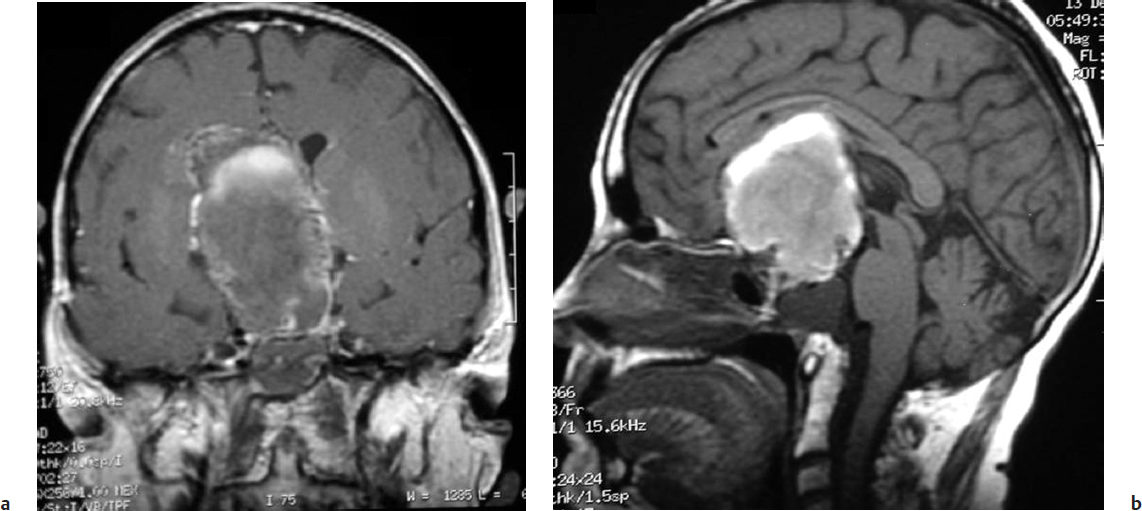
Fig. 35.3a,b (a) Coronal and (b) sagittal magnetic resonance imaging (MRI) of the same patient shown in Fig. 35.2. MRI demonstrated a 5 × 2 × 4 cm suprasellar mass with sellar extension and a largely cystic component. The patient underwent a right frontotemporal orbitozygomatic craniotomy for resection of tumor. A total resection was achieved, and the patient showed no signs of clinical or imaging recurrence 30 months later.
On imaging, craniopharyngiomas may be described in relation to the optic chiasm; craniopharyngiomas are characterized as prechiasmatic, retrochiasmatic, and subchiasmatic. Prechiasmatic tumors grow forward between the optic nerves, displacing the optic chiasm upward and backward as well as displacing the A-1 segment of the anterior cerebral artery.2 In contrast, retrochiasmatic craniopharyngiomas displace the chiasm forward and have a propensity to fill the third ventricle, resulting in obstructive hydrocephalus. With posterior and inferior extension, retrochiasmatic tumors may displace the basilar artery.2
With the introduction of intraoperative imaging modalities such as intraoperative MRI, the rate of gross total resection of difficult cases has been improved. Intraoperative MRI was used during surgery in 25 selected patients in whom tumor resection was anticipated to be difficult according to preoperative findings22; the rate of total tumor extirpation was increased by 16% (four additional patients) after reintervention based on preoperative MRI findings of residual tumor, leading to an 80% rate of gross total resection in the entire series. The rate of new ophthalmologic and endocrine deficits was acceptable compared with previously reported rates.22
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


