Abstract
With a prevalence of one in 3000–4000 newborns, congenital hypothyroidism (CH) is the most common inborn endocrine disorder and one of the most common preventable causes of mental retardation. While most cases are sporadic and associated with abnormalities of thyroid gland development and migration (thyroid dysgenesis), approximately 15–20% are caused by inherited defects in one of the steps of thyroid hormone synthesis (thyroid dyshormonogenesis). When the synthesis defect results in reduced hormone secretion, the ensuing diminished negative feedback on the anterior pituitary thyrotrophs leads to an increase in thyrotropin (TSH) secretion stimulating the thyroid gland. Consequently, patients are born with an enlarged thyroid gland (goiter) or develop goiter postnatally, especially when diagnosis and treatment with levothyroxine (L-T 4 ) are delayed.
Keywords
goiter, thyroid, congenital hypothyroidism (CH), levothyroxine (L-T 4 ), thyroxine (T 4 ), triiodothyronine (T 3 ), perchlorate (ClO 4 2 ) discharge test, hormonogenesis, dyshormonogenesis
Introduction
With a prevalence of one in 3,000–4,000 newborns, congenital hypothyroidism (CH) is the most common inborn endocrine disorder and one of the most common preventable causes of mental retardation. While most cases are sporadic and associated with abnormalities of thyroid gland development and migration (thyroid dysgenesis), approximately 15–20% are caused by inherited defects in one of the steps of thyroid hormone synthesis (thyroid dyshormonogenesis) ( Fig. 7.1 ). When the synthesis defect results in reduced hormone secretion, the ensuing diminished negative feedback on the anterior pituitary thyrotrophs leads to an increase in thyrotropin (TSH) secretion stimulating the thyroid gland. Consequently, patients are born with an enlarged thyroid gland (goiter) or develop goiter postnatally, especially when diagnosis and treatment with levothyroxine (L-T 4 ) are delayed. With the exceptions indicated below, these defects are inherited in an autosomal recessive fashion and are amenable to detection by newborn screening for CH.
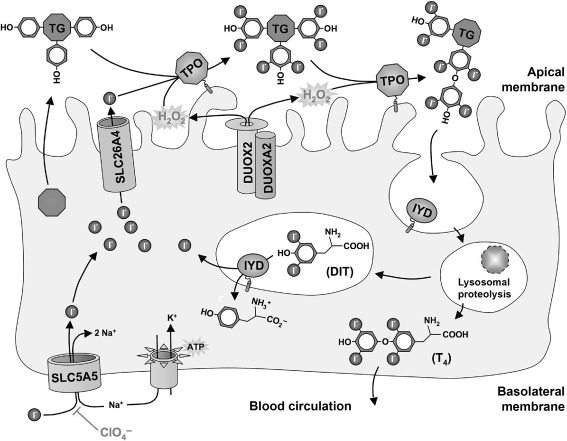
The etiological classification of CH is based on clinical and biochemical evaluation. Useful tests are measurement of serum TSH, thyroxine (T 4 ), triiodothyronine (T 3 ), and thyroglobulin [TG]; thyroid ultrasound and scintigraphy, using 99m Tc or, preferably, 123 I and, when indicated, the perchlorate (ClO 4 2 ) discharge test. Often infants with CH, confirmed by the TSH and T 4 values, are started on thyroid hormone replacement without detailed etiologic diagnosis. The latter is relegated to later years, usually two or three following a one-month withdrawal of L-T 4 replacement.
With the identification of the key steps involved in thyroid hormone synthesis, a molecular genetic diagnosis should be feasible for the vast majority of patients with dyshormonogenesis. A complete diagnostic work-up facilitates the selection of the most likely candidate gene(s) for etiologic confirmation. While the distinguishing features of each particular hormone synthesis defect are outlined in the following section, it should be noted that early genetic screening might be justified even without complete etiologic classification. Other than providing useful information for genetic counseling, there are additional benefits to a definite genetic diagnosis. One is the potential impact on treatment. For instance, patients with specific defects may be efficiently treated with iodide supplementation rather than L-T 4 . Another benefit is in the identification of a subset of patients with transient CH due to partial hormonogenesis defects. Even if euthyroid at a particular point in time, increased demand for thyroid hormone synthesis may precipitate hypothyroidism. An early molecular diagnosis predicts the necessity for lifelong hormone replacement therapy. Finally, some genetic defects may not manifest at birth but produce hypothyroidism later in life. The definitive diagnosis of an index case will enable early identification of subsequent cases in the same family and help to avoid the negative consequences on mental development associated with delayed diagnosis and treatment of hypothyroidism.
Pathophysiology and genetics of specific dyshormonogenesis defects
Defect in Thyroidal Iodide Trapping – Gene: SLC5A5 (NIS)
The sodium-iodide symporter (NIS; official gene symbol: SLC5A5 ) is a 13-transmembrane domain glycoprotein that mediates the uptake of iodide through the basolateral membrane of follicular thyroid cells ( Fig. 7.1 ). Iodide is actively concentrated into these cells by the electrogenic symport of Na + (two Na + for one I 2 ) down the electrochemical gradient maintained by the activity of Na + /K + -ATPase. NIS is also expressed, but not regulated, by TSH in several other differentiated epithelia, notably salivary glands, lachrymal glands, gastric mucosa, choroid plexus, and lactating mammary glands. All these tissues can concentrate iodide, but do not retain it because they lack the ability to bind it to a protein matrix.
The first congenital iodide transport or trapping defect (ITD) was found in a child, born to consanguineous parents, with an inability to concentrate iodide in the thyroid, salivary glands, and gastric mucosa. In 1997, a mutation in the gene-encoding NIS ( SLC5A5 ) was found to cause autosomal recessive ITD. The incidence is probably very low, given the quite characteristic clinical findings in iodide trapping defects and the small number of reported mutations. It should be noted, however, that unless TG is measured, the lack of thyroidal iodide uptake could lead to the erroneous diagnosis of athyreosis. Since 1997, 15 families (eight Japanese, two Brazilian, one Hispanic-Mexican, one Hutterite, one Spanish, one Italian, Argentinian, and one African from the Cameroon) with IDT have been genetically studied. Affected individuals were found to be homo- or compound heterozygous for 13 different SLC5A5 inactivating mutations (V59E, G93R, R124H, Q267E, C272X, G395R, T354P, fs515X, Y531X, ΔM143-Q323, ΔA439-P443, G543E, and g.-54C > T ). The age of onset of hypothyroidism is variable (neonatal, infancy, childhood) and seems to correlate with the residual in vitro activity of the mutant NIS.
Scintigraphy reveals blunted or absent radioactive iodide uptake (RAIU) in a normally located thyroid gland. The RAIU is a direct measure for NIS activity in vivo . RAIU in ITD is 0–5% (normal 10–40%) and imaging may suggest an absent thyroid gland (apparent athyreosis), especially when goiter is not prominent. Since the loss of NIS function is generalized, it also involves reduced salivary glands and gastric parietal cell uptake of iodide. Hence, there is little or no early 123 I or 99m TcO 4 − uptake resulting in the absence of salivary glands or stomach images during scintillation scanning. A simple and reliable test is the measurement of radioactivity in equal volumes of saliva and plasma obtained one h after the oral administration of 5 μCi of 125 I. A salivary-to-plasma ratio close to unity (normal 20) is pathognomonic of an NIS defect. The presence of TSH receptor blocking antibodies acquired transplacentally from a mother with autoimmune thyroid disease or inactivating mutations in the TSH receptor (causing resistance to TSH) will not affect the test or salivary gland and stomach imaging. Ultrasound examination further helps to distinguish defects in SLC5A5 from other conditions with reduced iodide uptake as it shows, characteristically, a normally located enlarged thyroid gland, excluding athyreosis and thyroid gland hypoplasia due to TSH receptor defects.
Although mutations in SLC5A5 appear to be a rare cause of dyshormonogenesis, genetic screening should be considered in all patients with permanent goitrous hypothyroidism in conjunction with low or absent RAIU. Without RAIU results, patients with goitrous hypothyroidism associated with high serum TG are better first evaluated for defects in TPO and DUOX2 . Apart from establishing a definite diagnosis, finding SLC5A5 mutations has further implications. Identification of an index case will allow subsequent prenatal diagnoses of other cases in the same family. The latter is critical, since patients with delayed onset of CH in ITD already had signs of developmental delay at time of diagnosis. Concerning treatment options, iodide supplementation can improve thyroid function in patients with residual NIS activity and should be considered, either alone or as adjunctive therapy together with L-T 4 replacement.
Defect in Efflux of Iodide Across the Apical Thyroid Cell Membrane – Gene: SLC26A4 (PDS)
SLC26A4 is a member of the multifunctional SLC26 transporter family facilitating the passive efflux of iodide across the apical thyrocyte membrane into the follicular lumen ( Fig. 7.1 ). In the inner ear, chloride/bicarbonate exchange by SLC26A4 is crucial for acid–base homeostasis of the endolymphatic fluid.
Biallelic inactivating mutations of SLC26A4 are a cause of Pendred syndrome (PDS). First described in 1896, it is clinically defined by congenital bilateral sensorineural hearing loss (associated with vestibular dysfunction) combined with diffuse or multinodular goiter. The latter usually develops in late childhood or early adolescence and is the consequence of a partial iodide organification defect consistent with the function of SLC26A4 as an apical iodide efflux channel in thyrocytes. However, about half of patients with SLC26A4 defects do not manifest thyroid abnormalities (designated nonsyndromic hearing loss with familial enlarged vestibular aqueduct or DFNB4). Nutritional iodide intake is an important modifier of the thyroid phenotype in PDS. High iodide intake may even completely prevent thyroid enlargement. With sufficient iodide in their diet, about 90% of patients are clinically and biochemically euthyroid. In the remaining 10% with elevated TSH level, goiter is always present.
Based on data for an English population, the incidence of SLC26A4 mutations causing isolated hearing loss or complete PDS can be estimated at 1:60,000. Note that in a substantial number of patients with clinically suspected PDS no SLC26A4 mutations were found, presumably because of genetic heterogeneity and phenocopies. Both goiter (particularly in iodine deficient regions) and congenital hearing loss of other causes are quite common.
PDS is rarely detected by neonatal screening for congenital hypothyroidism. Rather, patients present because of severe to profound congenital deafness. The finding of thyroid enlargement or a family history indicative of PDS would justify screening for SLC26A4 mutations. In the absence of goiter, children with suspected autosomal recessive nonsyndromic hearing loss should first be evaluated for mutations in the GJB2 (encoding gap junction connexin 26) gene, which account for up to 50% of all cases ( SLC26A4 mutations: up to 4%). In the case of negative GJB2 screening, either computed tomography or magnetic resonance imaging of the temporal bones should be considered. About 80% of patients with bilateral dilatation of the vestibular aqueduct and/or Mondini dysplasia were subsequently shown to have SLC26A4 mutations. There seems to be little additional diagnostic value in performing a perchlorate discharge test, which has a relatively high false negative rate (5%) in demonstrating partial iodide organification defect (PIOD; 10–90% radioiodide discharge) in patients with SLC26A4 mutations. The perchlorate (ClO 4 2 ) discharge test is based on the following physiological and pathologic considerations. Iodide transported into the follicular lumen is immediately covalently bound to TG (organified) and therefore does not normally require the concentrating activity of NIS for its retention. The test involves the administration of radioiodide, the uptake of which is measured by counting over the neck using a Geiger counter. Two hours later, ClO 4 2 is given, which blocks further iodide uptake by competitive inhibition of NIS. While bound iodine is retained, any inorganic iodide remaining in the thyroid gland is discharged and detected over the ensuing hour by falling counts over the gland. This occurs only if there is an organification defect, as in PDS, or other causes affecting protein binding of iodide.
Before systematic mutation scanning, targeted screening for the most common, recurrent mutations can be considered. L236P, T416P, and IVS8+1G≥A account for 50% of known SLC26A4 mutations in Caucasians of northern European descent, whereas H723R represents 53% of reported mutant alleles among Japanese. It has been suggested that, in the absence of a direct functional assessment in vitro , the pathogenicity of novel SLC26A4 mutations can be confidently predicted in cases of addition or omission of proline or a charged amino acid.
Defect in the Follicular Matrix Protein Providing Tyrosyl Groups for Iodide Organification – Gene: TG
Thyroglobulin (TG), a glycoprotein homodimer of 660 kDa, is the most abundantly expressed protein in the thyroid gland. It is secreted into the follicular lumen where it functions as matrix for hormone synthesis providing tyrosyl groups, the noniodide component of thyroid hormone ( Fig. 7.1 ). Iodinated TG constitutes the storage pool for thyroid hormone and iodide.
Although the existence of congenital TG defects was known in 1959, a demonstration of TG gene defect was first reported in 1991. Since that report, at least 40 distinct inactivating TG gene mutations have been described (see Ref. for a recent list of published mutations). Defects in TG as a cause of CH have been most extensively studied in Japanese subjects, for whom the estimated incidence is 1:67,000 equivalent to one-quarter to one-third of all cases with thyroid dyshormonogenesis.
Patients with biallelic TG gene defects typically manifest elevated serum TSH levels detectable on neonatal screening. As in other forms of dyshormonogenesis, free T 3 levels are usually disproportionately high compared to the low free T 4 serum concentrations, which have been explained by an increased intrathyroidal type 2 iodothyronine deiodinase activity converting T 4 to T 3 . Mild TG secretion defects can manifest as compensated hypothyroidism (isolated hyperthyrotropinemia). Goiters are often remarkably large, although early treatment of mild TG defects would prevent goitrogenesis. More revealing, serum TG is undetectable or very low in relation to the elevated serum TSH. Scintigraphy shows high uptake (due to induction of NIS expression by TSH stimulation) in a typically enlarged thyroid gland. Since the iodide organification process is not affected, there is, usually, no discharge after administration of ClO 4 2 . In the absence of TG, iodide is covalently bound to other proteins, such as albumin. Collectively, screening for TG gene mutations may be justified when an absent or unexpectedly low serum TG level is found in a CH patient presenting with normal-sized or enlarged thyroid gland.
Defects in the Enzymes Required for Iodide Organification
Defect in the Key Enzyme Catalyzing the Iodination and Coupling of Tyrosyl Moieties – Gene: TPO
Thyroid peroxidase (TPO) is a thyroid-specific heme peroxidase anchored via a C-terminal transmembrane domain at the apical membrane surface of follicular thyroid cells ( Fig. 7.1 ). Using hydrogen peroxide as oxidative equivalents, TPO catalyzes the iodination of tyrosyl residues in TG and the subsequent phenoxy ether bond formation between pairs of iodotyrosines to generate iodothyronines (T 4 and, to a lesser degree, T 3 and reverse T 3 ).
The first case of CH due to failure of iodide organification in the presence of hydrogen peroxide was reported in 1950 by Stanbury et al. Four decades later, the human TPO gene was cloned, followed shortly after by the description of a TPO mutation in a patient with CH. Inactivating biallelic defects in the TPO gene appear to be the most frequent cause of inherited dyshormonogenesis with permanent CH and the culprit in essentially all patients with permanent total iodide organification defects (TIOD; ≥90% ClO 4 − discharge). Bakker et al. estimated the incidence of TIOD owing to biallelic TPO defects at 1:66,000 for a Dutch population.
In about 20% of cases with TIOD, only monoallelic defects of TPO are found, presumably due to unidentified cryptic mutations in unexamined intronic or regulatory regions of the gene. Indeed, in a patient with TIOD and single allele mutation, TPO mRNA analysis of thyroid tissue obtained at surgery revealed monoallelic expression of only the mutant allele, indicating an unidentified defect on the other allele. Although heterozygous TPO mutations do not directly result in abnormal thyroid function, such monoallelic defects may play a role as genetic susceptibility factors in transient hypothyroidism. In a Chinese population, heterozygosity for a common TPO founder mutation (2268insT) is 16 times more common in babies with transient neonatal hypothyroidism compared to normal babies.
TPO is the indisputable candidate gene in patients with permanent TIOD. In contrast, nonsyndromic PIOD is heterogeneous, including defects in the hydrogen peroxide generator driving TPO activity. When the ClO 4 − discharge test is not available, screening for TPO mutations is still reasonable. For instance, 10 of 53 unrelated patients from Portugal with permanent CH, orthotopic thyroid gland, and elevated serum TG levels were found to harbor TPO defects. In an Eastern European population with an unusually high frequency of dyshormonogenesis (34% of CH), 18 of 39 apparently unrelated patients with permanent CH, normal or enlarged gland in situ , and normal or high serum TG were found to have TPO mutations. In 12 patients, only a single heterozygous mutation (1273_1276dupGGCC) was detected.
Finding of TPO mutations in a neonate with CH indicates that the patient will require lifelong treatment with thyroid hormone and that future pregnancies should be carefully monitored for the presence of fetal goiter. The latter can be detected by ultrasonography and treated by a single intraamniotic injection of L-T 4 to prevent goiter-related dystocia and improve neurological development.
Defect in the NADPH-oxidase Providing Hydrogen Peroxide for TPO – Gene: DUOX2 ( THOX2 )
Dual oxidases (DUOX1 and DUOX2; formerly known as thyroid oxidases or THOX) are NADPH oxidases expressed at the apical membrane of follicular thyroid cells ( Fig. 7.1 ). They provide hydrogen peroxide, the essential electron acceptor for the TPO-catalyzed iodination and coupling reactions. DUOX2 is also expressed at high levels in other epithelia, particularly in the gastrointestinal tract and salivary glands, and is proposed to function in a host defense mechanism.
Since the 2002 description of DUOX2 mutations in patients with CH, 26 different mutations have been reported. About half of these are nonsense, frameshift or splice site mutations predicting a dysfunctional enzyme lacking the C-terminal NADPH oxidase domain (G201fs, E327X, W414X, Y425X, R434X, L479fs, G488R, K530X, K628fs, Q686X, R701X, R842X, S965fs, Q1023X, Q1026X, g.IVS19-2A≥C). Of the missense mutations (Q36H, Y475C, A649E, H678R, E879K, R885Q, R110Q, D506N, R376W, G1518S), three have been studied in vitro and shown to cause either a complete or partial defect in trafficking of DUOX to the cell surface or reduced expression of an inactive protein.
Although most dyshormonogenesis defects are inherited in an autosomal recessive fashion, a single defective DUOX2 allele suffices to cause CH. When these patients were reevaluated at three years of age after withdrawal of L-T 4 , they often had normal thyroid function tests, indicating that the CH was transient. In addition, adult heterozygotes in these and other families with DUOX2 gene defects all had normal serum TSH concentrations. Since no evidence was found for dominant negative effects of the mutant DUOX2 proteins, these patients appear to have DUOX2 haploinsufficiency with manifestation limited to the neonatal period when thyroid hormone synthesis requirements are highest (from about 10 μg T 4 /kg/day progressively decreasing to around 3 μg T 4 /kg/day after the first year of life).
Several studies have linked biallelic DUOX2 defects to permanent CH with PIOD. Based on the type of mutations (nonsense, frameshift) or the in vitro study of missense mutations found in a homozygous or compound heterozygous state in these patients, most are predicted to express no residual DUOX2 activity. In contrast to the complete inactivation of TPO, which consistently leads to TIOD, a complete loss of DUOX2 activity does not completely abrogate the ability to synthesize the thyroid hormone. Of all the patients with biallelic DUOX2 defects, only one reportedly had TIOD. However, the results of the ClO 4 − discharge test in this patient are doubtful since L-T 4 treatment had not been discontinued. Indeed, a recent case report from Japan describes several patients with complete loss of DUOX2 activity due to biallelic frameshift mutations, who all presented with only transient CH and normal thyroid function tests in childhood. Limited iodide organification in these patients is likely maintained by the activity of DUOX1, which is also expressed in thyrocytes, albeit at a lower level compared to DUOX2.
With the increasing number of reported cases, phenotype–genotype correlations have become more complex than initially anticipated. The expressivity of DUOX2 defects is likely influenced by genetic background (e.g., DUOX1 ) and may, at least in part, depend on the iodide intake. Since iodination by TPO requires both iodide and hydrogen peroxide, a diet containing excessive amounts of iodide, common in Japan, would lead to better utilization of hydrogen peroxide provided by DUOX1. Further credence to an important role of iodide intake in expressivity of DUOX2 defects is provided by anecdotal reports showing that perinatal iodine overload completely normalized TSH levels in the early postnatal period, indicating compensation of the defect. This is opposite to the situation in normal infants in whom such iodine overload induces a physiological organification blockade (Wolff–Chaikoff effect).
The incidence of DUOX2 mutations in CH has not been determined. Certainly, DUOX2 mutations are frequent in patients with PIOD. For instance, seven of 20 such unrelated patients from Italy were found to have pathogenetic DUOX2 mutations. Screening of DUOX2 is therefore recommended in patients with nonsyndromic PIOD. In those with suspected nonsyndromic organification defect (normal sized to enlarged thyroid gland with high serum TG concentration) not confirmed by ClO 4 − discharge test, TPO should be screened first, especially in cases where CH is profound. Concerning follow-up, patients with transient CH due to DUOX2 haploinsufficiency are likely at risk for recurrent hypothyroidism at times of increased hormonogenesis requirements, such as pregnancy. With regard to treatment after the neonatal period, we would advocate assessment of iodide supplementation as an alternative to potentially life-long L-T 4 replacement therapy.
Defect in the DUOX2 Cofactor – Gene: DUOXA2
Two novel genes, called DUOX maturation factors ( DUOXA1 and DUOXA2 ), were recently identified in the DUOX1 / DUOX2 intergenic region. These genes are oriented head-to-head to the DUOX genes and thus form bidirectional transcriptional units with their corresponding dual oxidase genes. This arrangement ensures coexpression of DUOXA2 with DUOX2 (and DUOXA1 with DUOX1 ). The DUOXA genes encode integral membrane proteins essential for the endoplasmic reticulum-to Golgi transition, maturation, and translocation to the plasma membrane of functional DUOX enzymes ( Fig. 7.1 ).
In 2008, the first mutation in DUOXA2 was described in a Chinese patient with PIOD and mild, permanent CH. The patient was homozygous for an Y246X nonsense mutation that resulted in a complete loss of DUOXA2 function in vitro . A heterozygous Y246X carrier was also identified among 92 unrelated Han Chinese control individuals suggesting that this mutation could be relatively common in this population. In fact, a recent report described another Chinese CH patient with gland in situ that was compound heterozygous for Y246X and another nonsense variant, Y138X. In a patient with mild CH of European descent, Hulur et al. identified a DUOXA2 missense mutation (C189R) that abolished the functional expression of the protein in vitro . This patient was again a compound heterozygote, harboring a large deletion of the DUOX2/DUOXA2/DUOXA1 region on the paternal allele. In all three reported cases, the loss of a single DUOXA2 allele did not lead to abnormal thyroid function, in contrast to the haploinsufficiency caused by monoallelic DUOX2 mutations. Apart from the intact DUOXA1/DUOX1 system, an additional mechanism for maintaining adequate hydrogen peroxide supply in patients with DUOXA2 deficiency is the partial activation of DUOX2 by DUOXA1, as demonstrated in vitro . Since DUOXA2 defects lead to secondary deficiency of functional DUOX2 enzyme, one can anticipate that expressivity will be similarly modulated by nutritional iodide as described for DUOX2 defects.
Defect in Iodide Recycling with Secondary Iodide Deficiency – Gene: IYD ( DEHAL1 )
The lysosomal proteolysis of endocytosed iodinated TG liberates the iodothyronines (T 4 ≥ T 3 ). However, most iodide contained in TG is released as uncoupled mono- and diiodotyrosines (MIT, DIT). MIT and DIT are subject to NADPH-dependent reductive deiodination by iodotyrosine deiodinase (IYD, or dehalogenase) leading to formation of free iodide and tyrosine, both of which can be reutilized in hormone synthesis ( Fig. 7.1 ).
IYD contains an N-terminal membrane anchor, a less conserved intermediate domain, and a C-terminal domain resembling enzymes of the bacterial NADH oxidase/flavin reductase superfamily. The protein is predominantly localized at the apical thyroid cell membrane and in subapical, endosomal compartments, with the catalytic domain facing outside the cell or into the endosomal lumen, respectively. In addition to the thyrocyte, the enzyme is expressed in liver and kidney. The expression in the latter tissue serves for the execution of a pathognomonic in vivo test.
A congenital defect in iodotyrosine deiodination was first described in 1953 in a consanguineous group of Scottish itinerant thinkers. In 2008, molecular defects in IYD underlying impaired intrathyroidal dehalogenation were described in four unrelated consanguineous families. Six affected individuals were homozygous for either a missense mutation (R101W, I116T, or A220T) or a combined missense/deletion mutation c.315delCAT (resulting in replacement of both F105 and I106 by leucine at position 105). All mutations map to the flavin-binding domain and virtually abolished the capacity of IYD to dehalogenate MIT and DIT in vitro . Notably, one heterozygous carrier of A220T developed nonautoimmune goitrous hypothyroidism at 15 years of age, pointing to a possible dominant behavior of the mutation in some individuals.
Loss of IYD activity prevents the normal intrathyroidal iodide “recycling” and leads to excessive urinary secretion of MIT and DIT. Since the resulting iodide deficiency does not manifest at birth, patients with biallelic IYD mutations tested normal at neonatal screening for CH. They subsequently came to medical attention at 1.5–8 years of age because of sequelae of hypothyroidism. On scintigraphy, a very rapid and high initial uptake of 123I in the enlarged thyroid is observed, followed by a relatively rapid spontaneous decline of the accumulated iodine without the administration of ClO 4 2 . A pathognomonic finding is the intact excretion in urine of intravenously administered MIT or DIT, without removal of the iodine. The detection of high urinary MIT and DIT by tandem mass spectroscopy may become a useful diagnostic test.
The incidence of IYD mutations is unknown. Although not a viable candidate gene for CH, a potential role for IYD variants in susceptibility to endemic goiter remains to be investigated. We suggest that screening of IYD may be considered in patients developing “idiopathic” diffuse or multinodular goiter between the neonatal period and adolescence, while early and late images during 123I scintigraphy are compatible with IYD defects. However, deficient nutritional iodide intake, as in areas of endemic goiter, dietary goitrogens, and autoimmune thyroid disease has to be excluded. An autosomal recessive inheritance pattern of the disorder, as well as consanguinity of the parents, would likely increase the yield of mutation screening. Anecdotal evidence indicates that iodine supplementation (Lugol’s solution) is an effective alternative to L-T 4 treatment.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


