Fig. 2.1
The Complement Activation Pathways and its regulators. The Complement System: The classical pathway is initiated by immune complexes that activate C1. Activated C1 then recruits C4 and C2 to form a C3-convertase (C4b2a) that fragments C3 into C3a, an anaphylotoxin, and C3b, a molecule at the core of the complement system that binds covalently to hydroxyl groups on carbohydrates or proteins in bacterial surfaces and/or cell membranes. C3b tags invading microorganisms for opsonization and also amplifies the activation cascade acting as a focal site for further complement activation. The phylogenetically older alternative pathway is activated by low-grade cleavage of C3 in plasma; the resulting C3b binds factor B, a protein homologous to C2, to form the C3bB complex. Bound to C3b, factor B is cleaved by the activator factor D, which circulates in an active form as a serine esterase to form C3bBb: the C3-convertase of the alternative pathway. Because basal “tick over” deposition of C3b occurs in all cells exposed to complement, C3b is always available to prime the alternative pathway. However, continued activation and amplification is normally restricted because cell membranes do not bind factor B efficiently, and the inhibitory factor H prevents the association of factor B with C3b. Amplification of the alternative pathway occurs when factor H binds to foreign carbohydrates leaving factor B free to complex with C3b. Specificity for activation of the alternative pathway resides in the ability of factor H to discriminate between “self” and foreign carbohydrate determinants (Walport 2001a, b; Morgan 1995). The MBL pathway is initiated when the plasma mannose-binding lectin (MBL) protein, structurally similar to C1q (Matsushita et al. 1998), in a complex with the mannose-binding lectin-associated proteases 1 and 2 (MASP1 and MASP2) binds to an array of mannose groups on the surface of microorganisms. This binding activates MASPs, which then cleave C2 and C4 leading to the formation of the C3-convertase C4b2a in a manner comparable to activated C1 (Walport 2001a). Terminal complement pathway: The three complement activation pathways eventually converge at the level of C3 with formation of C3b and a C5 convertase that cleaves C5 into C5a, an anaphylotoxin, and C5b. This is the last enzymatic step of the activation cascades; thereafter, the complement pathways share a common sequence through the terminal components C6, C7, C8, and C9, leading to the generation of the membrane attack complex or MAC, the main effector of complement-mediated tissue damage. The terminal complement components – C6 through C9 -necessary to form the MAC, are constitutively present in plasma. Formation of the MAC is initiated by C5b followed by the sequential association of C6 into C5b6, and then C7, C8, and C9. C5b6 is a stable protein that binds to C7 to form the C5b-7 complex that inserts into the lipid bilayer of the plasma membrane. Membrane-bound C5b67 exposes a binding site for C8 leading to formation of the C5b-8 complex, which functions as a docking site for C9. A single C9 first binds to C8 in the C5b-8 complex; then C9 polymerizes forming the C5b-9 complex known as the membrane attack complex or MAC. The MAC is a circular polymer of variable number of C9 monomers (Podack 1984; Podack and Tschopp 1984; Podack et al. 1982; Podack and Tschopp 1982; Tschopp et al. 1984) with the capacity to insert into cell membranes (Laine and Esser 1989) and form a transmembrane pore with hydrophobic domains on the outside and hydrophilic domains in the inside, and an effective internal radius of 5–7 nm (Mayer 1984; Ramm et al. 1982, 1985). Restriction of complement activity by complement regulators: In the fluid phase, the C1 inhibitor regulates the classical pathway by targeting C1, factors H and I regulate the alternative pathway, and S-protein, clusterin and serum lipids compete with membrane lipids for reacting with nascent C5b67. In addition to these fluid phase inhibitors, several membrane proteins, decay-accelerating factor (DAF) (Nicholson-Weller et al. 1985), membrane cofactor (MCP) (Brooimans et al. 1992; Cervoni et al. 1992) and complement receptors 1and 2 (CR1, CR2) regulate the C3-convertases (Beynon et al. 1994) a major amplification step in the early complement activation cascade, while clusterin (McDonald and Nelsestuen 1997), S-protein (Podack and Muller-Eberhard 1978) and CD59 inhibit formation of the MAC (Fletcher et al. 1994; Bodian 1997; Davies et al. 1989; Huang et al. 2006; Husler et al. 1994; Meri et al. 1990; Petranka et al. 1992). Massive or unrestricted activation of complement and MAC formation leads to cell lysis, as seen in immune hemolytic anemias or PNH. Basal (tick over) activation with or without reduced restriction in nucleated cells leads to transient MAC pores formation, influx and efflux of ions and macromolecules, and activation of cellular pathways that contribute to proliferation, inflammation and thrombosis as characteristically seen in target organs of diabetic complications
2.2.1 Membrane Attack Complex (MAC)
The formation and function of MAC is described in Fig. 2.1. The MAC is a trans-membrane pore that allows influx of salt and water leading to colloidosmotic lysis of MAC-targeted cells. The formation of the MAC pore, however, seems to be the end stage of a process that transits through a reversible phase during which MAC pores can be formed transiently (Halperin et al. 1993, 1988; Acosta et al. 1996) generating significant changes in the membrane permeability and internal composition of MAC-targeted cells without compromising their viability (Halperin et al. 1993, 1989; Berger et al. 1993). Figure 2.1 illustrates the large increase of intracellular Ca++ levels caused by transient, non-lytic MAC pores on isolated guinea-pig cardiomyocytes (Berger et al. 1993). Thus, while the matured MAC pore can kill non-self cells by perforating their plasma membrane, the transient MAC pore can induce non-lethal biological responses in “self” cells, and mediate physiological and/or pathological response (Nicholson-Weller and Halperin 1993).
2.2.2 MAC Cellular Signaling and Complement Activation
The MAC is a mediator of cellular signaling and an effector of organ pathology. Activation products of complement such as C3b and C4b or the anaphylotoxins C3a and C5a exert their biological activities through binding to specific receptors expressed in many cell types. Upon binding to their cognate ligands, these receptors trigger signaling processes mostly linked to inflammation and immune responses (Carroll 1998). In contrast, there are no specific receptors for the MAC, which inserts directly into the lipid bilayer of the plasma membrane. Insertion of transient non-lethal MAC pores into cell membranes induces an array of biological responses that promote cell proliferation, inflammation and thrombosis, as characteristically seen in all diseases in which focal complement activation is a characteristic pathological abnormality. These responses are mediated by growth factors and cytokines including bFGF and interleukin-1 (Benzaquen et al. 1994; Acosta et al. 1996) which are directly released through the MAC pore, and PDGF (Benzaquen et al. 1994), monocyte chemotactic protein-1 (MCP-1) (Torzewski et al. 1996) and von Willebrand factor (Hattori et al. 1989) which are released through canonical secretion pathways in response to intracellular perturbations induced by the MAC (See Fig. 2.2). Acting auto- or para-crinically on extracellular receptors, these leaked/secreted molecules promote (a) proliferation of fibroblasts, endothelial and smooth muscle cells (Benzaquen et al. 1994), (b) inflammation through the expression of pro-inflammatory adhesion molecules such as P-selectin, VCAM-I and E-selectin (Marks et al. 1989; Libby 2002), and the attraction of monocytes and macrophages to the site of focal complement activation (Kostner 2004) and (c) thrombosis through the expression of pro-thrombotic tissue factor, and the transient assembly of the prothrombinase complex in both the endothelial cell surface and microvesicles shed from their plasma membrane (Hamilton et al. 1990; Christiansen et al. 1997; Saadi et al. 1995). Our group has also shown that the MAC promotes in vitro accumulation of cholesteryl esters into mouse macrophages fostering their conversion into foam cells (Wu et al. 2009a).
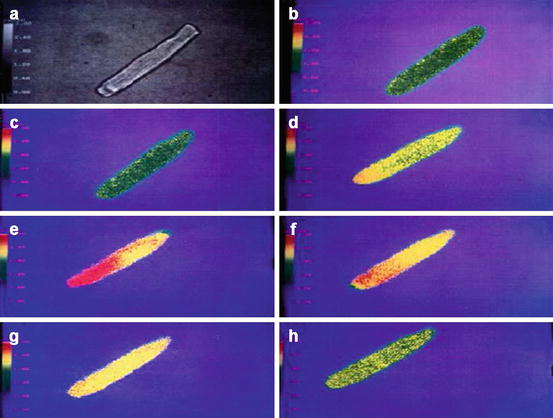
Fig. 2.2
Non-lytic transient MAC triggers a large increase of intracellular Ca++ concentration ([Ca ++ ] i ). The figure shows pseudo-color images of isolated adult rat ventricular muscle cell loaded with fura-2, a fluorescent calcium-indicator dye. Increasing emission intensities were associated with increasing [Ca++]i, coded in a pseudo-color spectrum from blue (low Ca++) to red (high Ca++). (a) Reference, digitalized phase-contrast image. Before adding complement factors, the cell shows low emission intensities (yellow-green) (b) that does not significantly change after addition of C5b6 plus C7 and C8 (c). After addition of C9, the cell showed a marked emission increase (d–e), representing an increase in [Ca++]i that lasted ≈1 min. After an additional 3 min, [Ca++]i decreased to near baseline levels (f–h) (Figure reproduced from Berger et al. 1993)
Reversible insertion of transient MAC into cell membranes also activates intracellular signaling pathways (Tegla et al. 2011) including increased (a) Ca++ influx (Acosta et al. 1996; Morgan and Campbell 1985) and Ca++-activated K+ efflux (Halperin et al. 1989) (b) production of reactive oxygen species (ROS) (Adler et al. 1986; Bellosillo et al. 2001), (c) activation of Ca++-sensitive and Ca++-insensitive protein kinase C (PKC) (Cybulsky et al. 1990; Carney et al. 1990), (d) activation of heterotrimeric G proteins of the Gi/Go subfamily (Niculescu et al. 1994), (e) mitotic signaling through the small G-protein Ras, that induces Raf-1 translocation triggering activation of the ERK pathway (Niculescu et al. 1997), and (f) activation phosphatidylinositol-3 kinase (PI3-K) (Niculescu and Rus 2001). The MAC also activates the transcription factor NF-κB, which induces the production of IL-6 as well as IL-8 and MCP-1 (Kilgore et al. 1997; Viedt et al. 2000). In glomerular mesangial cells, insertion of the MAC results in increased production of ROS (Adler et al. 1986) and the release and/or up-regulation of (a) bFGF and PDGF (Benzaquen et al. 1994) which induce mesangial cell proliferation and transcriptional up-regulation of TGF-β (Torbohm et al. 1990; Lovett et al. 1987); (b) monocyte chemotactic protein-1 (MCP-1), which attracts monocytes and macrophages to the mesangium (Stahl et al. 1993); and (c) transcriptional activation of collagen type IV (Wagner et al. 1994).
Important in the context of this chapter, many mediators and cellular signaling pathways activated by the MAC are established players in the tissue damage seen in the target organs of diabetic complications (Kotajima et al. 2000; Adler et al. 2000) and are common to or intersect with complement-independent mechanisms up-regulated by hyperglycemia such as overproduction of ROS, up-regulation of PKC and NF-κB (Brownlee 2001, 2005; Giacco and Brownlee 2010).
2.2.3 Complement Regulation Through CD59
Given the potentially devastating effects of uncontrolled complement activation, it is not surprising that an array of mechanisms have evolved to protect “self” cells from complement attack and MAC formation. On the one hand, the inherent instability of the enzymes in the activation pathways and the extremely short half-life of the activated complement components coupled to the transient nature of the reaction sequences are intrinsic properties of the complement system that keep its activation restricted and limit the effects of complement to the vicinity of the activation site. On the other, a multitude of complement regulatory molecules exist that restrict critical steps of the pathways (See Fig. 2.1).
In particular, CD59 is a specific inhibitor of MAC formation that restricts C9 polymerization. Ubiquitously expressed in mammalian cells, CD59 is an 18–20 kDa glycosylphosphatidylinositol (GPI)-linked glycoprotein (Kooyman et al. 1995; Morgan 1995). CD59 inhibits the binding of C9 to C5b-8 by competing for binding to a nascent epitope on C8 thereby affecting the association of C9 to the C5b-8 complex (Ninomiya and Sims 1992). CD59 can not only function as an inhibitor of the formation of large MACs but can also allow cells to eliminate newly formed MACs by blocking the early, functional channel formation of MAC complexes (Kimberley et al. 2007).
Several lines of evidence indicate that CD59 is the most critical of the complement regulators in protecting human cells from MAC formation and MAC-induced phenomena: erythrocytes from a family carrying the Inab blood group phenotype totally lacked decay-accelerating factor (DAF), an inhibitor of early complement activation, without any associated hemolytic disease (Lin et al. 1988; Telen and Green 1989; Reid et al. 1991; Sun et al. 1999); in contrast, individuals carrying CD59 mutations leading to undetectable levels of CD59 on their cell membranes exhibited an early onset hemolytic phenotype associated with vascular disease or chronic relapsing polyneuropathy (Yamashina et al. 1990; Nevo et al. 2013).
The GPI anchor in GPI-anchored proteins is introduced as a post-translational modification mediated by a protein called Pig-A that is required for the synthesis of the GPI-lipid tail (Luzzatto and Bessler 1996; Miyata et al. 1993, 1994; Takeda et al. 1993). Deficiency of GPI-linked proteins including CD59 in erythrocytes and platelets is phenotypically expressed as a complement-mediated hemolytic anemia, known as paroxysmal nocturnal hemoglobinuria (PNH) (Halperin and Nicholson-Weller 1989; Parker 1996; Risitano 2012). This paradigmatic disease, together with the silencing CD59 mutations described above, illustrates how the delicate balance between complement activation and restriction can be perturbed by the functional deficiency of CD59, leading to a potentially harmful pathological response even in the absence of complement-activating stressors.
2.3 Clinical Evidence for a Role of Complement in the Pathogenesis of Diabetes Complications
Evidence for a potential role of complement in the pathogenesis of diabetes complications was initially provided by the identification of activated complement proteins in target organs of diabetes complications biopsied or excised from individuals with diabetes (Rosoklija et al. 2000; Zhang et al. 2002; Falk et al. 1983a, b, 1987).
2.3.1 Diabetic Nephropathy
Falk et al. used antibodies against activated complement proteins including a neoantigen of the C9 portion the MAC to perform immunoelectron (IE) and immunofluorescent (IF) microscopy on kidney tissue from normal humans and individuals with insulin-dependent diabetes (Falk et al. 1983a, b, 1987). Staining of tissues with specific antibodies directed against the MAC neoantigen is the most direct method for determining whether complement activation has occurred to completion because the neoantigen is exposed only when the MAC is fully assembled and inserted into a cell membrane. IF microscopy of kidney tissue from individuals with insulin-dependent diabetes and varying degrees of mesangial expansion and glomerulosclerosis demonstrated a positive association between the degree of tissue damage and the amount of MAC deposited in the mesangium. IE microscopy of specimens from individuals with diabetic nephropathy revealed that the anti-MAC neoantigen monoclonal antibody reacted with linear and circular membranous structures within the mesangium, tubular basement membranes, blood vessel walls, and the glomerular basement membranes (Falk et al. 1983a, b, 1987). Our own studies confirmed glomerular MAC deposition (co-localized with GCD59, Fig. 2.4b) in kidney biopsies from individuals with diabetes (Qin et al. 2004). We also reported the presence of extensive MAC deposition in glomeruli and blood vessels in the biopsy of a kidney transplanted 2 years earlier and diagnosed as “recurrent diabetic nephropathy with no signs of rejection” (Qin et al. 2004).
Also, several studies have found that serum concentrations of mannose-binding lectin (MBL), an indicator of the lectin complement pathway, were significantly elevated in patients with type 1 diabetes (Bouwman et al. 2005; Hansen et al. 2003) and even more elevated in patients with vascular complications including diabetic nephropathy (Hansen et al. 2004, 2010; Saraheimo et al. 2005). Further, the FinnDiane Study showed that the baseline levels of MBL were positively associated with the progression and prognosis of diabetic nephropathy in type-1 diabetes (Hovind et al. 2005; Kaunisto et al. 2009). Transcriptome analyses of human kidneys have shown that the canonical complement-signaling pathway is differentially up-regulated in both glomeruli and tubules of individuals with diabetic nephropathy and is associated with increased glomerulosclerosis. Woroniecka et al. showed that increased expression of C3 mRNA and protein in glomeruli was positively correlated with diabetic kidney disease (Woroniecka et al. 2011). C3 is a key protein central to all complement activation pathways.
2.3.2 Diabetic Retinopathy
A combination of biochemical, histological and genetic studies suggest that aberrant function of the complement system plays a key role in the etiology of age-related macular degeneration, a common degenerative ocular disease (Wlazlo et al. 2014). Two independent studies have documented that complement activation occurred extensively and to completion in the choriocapillaries of eyes from donors with diabetic retinopathy, while similar deposits were not found in eyes from donors without retinopathy. In the study by Gerl et al., intense positive staining for MAC deposits (neoantigens) and C3d was noted without exception in all 50 eyes with diabetic retinopathy analyzed but only in one of 26 eye donors without diabetic retinopathy (Zhang et al. 2002; Gerl et al. 2002).
2.3.3 Diabetic Neuropathy
Nerve damage through complement activation has been reported in several peripheral neuropathies (Kaida and Kusunoki 2010). Interestingly, a recent study showed that a cell surface deficiency of the complement regulatory protein CD59 due to a single missense mutation of the CD59 gene was clinically expressed by a form of chronic peripheral neuropathy in children (Nevo et al. 2013). The presence of activated complement proteins and MAC neoantigen in sural nerve biopsies from individuals with diabetes was first reported by Hays and collaborators (Rosoklija et al. 2000); in the same nerve biopsies analyzed in that study, we observed co-localization of MAC and GCD59 (Qin et al. 2004).
2.3.4 Diabetic Cardiovascular Diseases
A relatively large body of evidence supports a role of complement in cardiovascular pathology in general and atherosclerosis in particular (Hertle et al. 2014; Kostner 2004; Wu et al. 2009a, b; Haskard et al. 2008; Niculescu and Rus 1999; Lappegard et al. 2014; Niculescu et al. 1987; Pang et al. 1979; Schmiedt et al. 1998; Seifert et al. 1989; Seifert and Kazatchkine 1988). Diabetes and pre-diabetes are major independent risk factor for cardiovascular disease (Tominaga et al. 1999; Jarrett et al. 1982). A potential role of C3, its activation products and other components of the early complement cascade in the pathogenesis of type 2 diabetes has been suggested from associations found in several cross-sectional and longitudinal human studies, as recently reviewed in (Hertle et al. 2014). Also, cleavage of C3 generates the anaphylatoxins, C3a and C5a, and further cleavage of C3a by carboxypeptidases leads to production of acylation stimulating protein (ASP/C3adesArg), a lipogenic hormone that plays a role in atherosclerosis (Onat et al. 2011; Hertle et al. 2012; Cianflone et al. 2003). However, the potential role of complement activation in diabetic cardiovascular disease has not been fully explored in human studies. In a sub-analysis of the DIGAMI-2 trial, which prospectively studied more than 1,200 individuals with type 2 diabetes who had a myocardial infarction, high blood levels of soluble MAC at the time of hospitalization strongly and independently predicted the risk of a cardiovascular event in the subsequent 3.5 years (Mellbin et al. 2012). In the same sub-analysis, levels of mannan-binding lectin serine peptidase 2 (MASP-2) appeared to be associated with a poor cardiovascular prognosis, although this did not remain significant after multivariable adjustments.
Together, the findings summarized above suggest an important role of the complement system in the development and progression of diabetic complications. However, it is important to note that whether the reported associations of elements of the complement system with diabetes represent causative pathogenic precursors or are just a collateral consequence of diabetes and its complications is still unclear and deserves further investigation. Complimentary to the clinical evidence discussed above, in vitro and animal studies seem to suggest that the lectin MBL pathway could be activated by increased levels of MBL induced by hyperglycemia and by a cooperative binding of MBL to fructosyllysine, the product of the non-enzymatic attachment of glucose to α- or ε-amino groups in lysines (Fortpied et al. 2010; Ostergaard et al. 2013).
2.4 Glycated CD59
2.4.1 Functional Inactivation of CD59 by Glycation
Glycation-inactivation of CD59 is a molecular link between complement regulation and the complications of diabetes. There are different ways in which the delicate balance between complement activation and restriction can be unfavorably perturbed in diabetes. On the one hand, autoantibodies to glycated and glycol-oxidized proteins can activate the classical pathway (Uesugi et al. 2004; Ostergaard et al. 2013; Orchard et al. 1999; Witztum et al. 1983), fructosamines, reportedly activate the lectin pathway by an increased ligand-receptor binding (Fortpied et al. 2010) and increase formation of advanced glycation end products (AGEs) on the arterial walls can serve as neo-epitopes for MBL binding (Flyvbjerg 2010). The presence of activated products of C3 (C3d and iC3b), C2, and C4 in target organs of diabetic complications (Gerl et al. 2002; Rooijakkers et al. 2009) is consistent with activation of complement through either or both of these two pathways. On the other hand, functional inhibition by glycation or reduced expression of complement regulatory proteins such as CD59 (Acosta et al. 2000; Zhang et al. 2002) could lead to increased complement-mediated damage and MAC-induced biological processes likely to contribute to the pathogenesis of diabetic complications (Rooijakkers et al. 2009; Zhang et al. 2002; Falk et al. 1983a, b, 1987; Weiss et al. 1990).
Of all the above-mentioned putative mechanisms that potentially contribute to complement-mediated tissue damage in diabetes, glycation-inactivation of CD59 is the most extensively studied and documented. Hereafter, we will summarize the experimental and clinical evidence for a role of GCD59 in the pathophysiology of diabetes complications and the potential utility of glycated CD59 as a diagnostic biomarker in diabetes.
Protein glycation on α- or ε-amino groups is a well-established mechanism of tissue damage in diabetes. Despite the many amino groups exposed on the surface of either soluble or membrane proteins, very few ε-amino groups are glycated to a significant extent. For example, despite the very close sequential proximity of lysyl residues K61, K65, and K66 in β-hemoglobin, only the ε-amino group of K66 is preferentially glycated in vivo (Shapiro et al. 1980). This is because significant ε-amino glycation requires effective acid-base catalysis, which occurs at “glycation motives” such as histidyl/lysyl residues in which the imidazolyl moiety is located at ≈5A° from the ε-amino group of the lysyl residue or at lysyl-lysyl pairs (Shapiro et al. 1980; Iberg and Fluckiger 1986; Shilton and Walton 1991; Shilton et al. 1993; Calvo et al. 1993; Flückiger and Strang 1995).
Analyzing the reported NMR structure of human CD59 (Fletcher et al. 1994), our group predicted that the protein contains within its active site a glycation-motif formed by amino acid residues K41 and H44 and that glycation could inhibit the activity of CD59 (See Fig. 2.3a). This idea was further supported by the fact that K41 is adjacent to W40, a conserved residue that is essential for CD59 function (Bodian 1997; Yu et al. 1997). Exposing purified or recombinant human CD59 to glucose, we demonstrated that the anti-MAC activity of CD59 is indeed inhibited by glycation, as documented with anti-glycated CD59 antibodies (See Fig. 2.3) (Acosta et al. 2000). Site-directed mutagenesis of human CD59 confirmed experimentally that (1) human CD59 is inhibited by glycation of its K41 residue and (2) H44 is required for glycation-inactivation of human CD59 (Acosta et al. 2000) (See Fig. 2.3b).
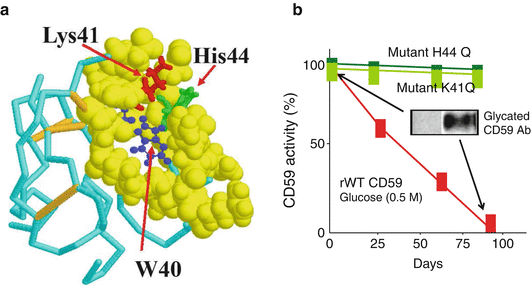
Fig. 2.3
Glycation of CD59 (a): NMR structure of human CD59 (From Fletcher et al 1994); the figure highlights the Lys41 (red) and His44 (green) amino acid residues that conform the glycation motif and the Trp40 (blue) residue, a conserved amino-acid critical for CD59 activity. The His44 of human CD59 is not found in CD59 from any other species sequenced to date. (b) Glycation inactivates human CD59: the activity of recombinant human CD59 was measured in a hemolytic assay using guinea-pig erythrocytes (GPE) exposed to human MAC formed with purified terminal complement components (as reported in Acosta et al. 2000). Incubation of CD59 in the presence of glucose progressively inhibits CD59 activity (red line). Inhibition of activity is associated with glycation of CD59, because (a) parallel incubation of the same preparation with the non-glycating sugar sorbitol did not affect CD59 activity (see Fig. 2 in Acosta et al. 2000). An antibody specific for the glycated form of CD59 showed the presence of glycated CD59 in the protein inactivated after incubation with glucose (inset). Site directed mutagenesis of either K41 or H44 generated two CD59 mutants (mutant K41Q and mutant H44Q) that were active against human MAC but not inhibited by exposure to glucose (Figure reproduced from Acosta et al. (2000))
2.4.2 Animal Studies
The significance of the glycation motif in human CD59 is highlighted by the fact that the residue H44 is absent in CD59 from any animal species sequenced to date (See Table 1 in Acosta et al. 2000). This is remarkable because humans are particularly prone to develop diabetic vascular complications, and it is well known that no single animal model of the disease can recapitulate the extensive vascular disease that commonly complicates human diabetes (Vischer 1999). This observation has led to the intriguing hypothesis that the presence of a glycation motif K41-H44 in human CD59 (not present in CD59 from other species) could contribute to the unique sensitivity of humans to develop extensive vascular disease in response to hyperglycemia. To test whether the functional inactivation of CD59 confers higher risk for diabetes-induced atherosclerosis, our group has generated molecular engineered mice that are completely deficient in CD59 (mouse CD59 knockout (mCD59KO) mice)), crossed them into the ApoE−/− background (one of the models recommended by the Diabetes Complications Consortium for studying diabetes-induced atherosclerosis (Hsueh et al. 2007)) and rendered them diabetic by injection of low doses of streptozotocin (an experimental model of type 1 diabetes also recommended by the Diabetes Complications Consortium (Brosius et al. 2009)). The ablation of CD59 (mCD59 knock out mice in an ApoE−/− background) promoted accelerated atherosclerosis with occlusive coronary disease, vulnerable plaque, and premature death (Wu et al. 2009a; An et al. 2009). In contrast, either transgenic overexpression of human CD59 in the endothelium or the administration of a neutralizing anti-C5 monoclonal antibody in these mice significantly attenuated the development of atherosclerosis (Morgan 1995). These observations in engineered mice are consistent with a body of experimental evidence supporting a role of complement in the development of atherosclerosis (Haskard et al. 2008; Niculescu and Rus 1999; Pang et al. 1979; Schmiedt et al. 1998; Buono et al. 2002; An et al. 2009). Diabetic mice: Initial evidence demonstrated that diabetic mCd59KO/Apoe −/− mice developed a severe and accelerated form of atherosclerosis characterized by significantly larger atherosclerotic lesion areas in the aorta (>2-fold increased lesion area in en-face aorta preparations) and the aortic roots, as compared to their non-diabetic littermates. This accelerated atherosclerosis observed in diabetic CD59 deficient mice was associated with significantly increased MAC deposition (detected with anti-mouse C9 antibodies) and occurred at levels of hyperglycemia, lipid profiles and body weights similar to those in diabetic CD59 expressing mice (Liu et al. 2014). Interestingly, diabetic mCD59KO mice had significantly increased MAC deposition and higher levels of serum iC3b, a byproduct of C3 cleavage during activation of complement, than non-diabetic mCD59KO mice. These two observations are consistent with the emerging evidence for activation of the complement system in diabetes (Flyvbjerg 2010; Uesugi et al. 2004; Fortpied et al. 2010; Orchard et al. 1999), as discussed above.
2.4.3 Functional Studies of Diabetics
The identification of the cellular and molecular mechanisms by which hyperglycemia causes tissue damage in humans has been hampered by (1) the lack of animal models that recapitulate the combination and intensity of complications seen in human diabetes (Vischer 1999), (2) the chronic nature of the complications that can take decades to become clinically evident, and (3) ethical and practical considerations that preclude conduction of the functional studies required to assess mechanistic hypotheses in humans. In spite of these limitations, two lines of human experimental evidence seem to support the potential role of glycation-inactivation of CD59 in the pathogenesis of diabetes complications.
Functional inactivation of CD59 was found in individuals with diabetes. If glycation inactivates CD59 in vivo, one would predict an increased sensitivity to MAC-mediated phenomena in cells/tissues carrying glycation-inactivated CD59. This prediction was confirmed in an ex-vivo study using red blood cells (RBC) from donors with or without diabetes in which we measured both the activity of CD59 in the RBC membranes and the RBC sensitivity to human MAC. The results showed that RBC from individuals with diabetes (selected to carry a HbA1c >8 %) exhibited a significantly reduced activity of CD59 and were significantly more sensitive to MAC-mediated lysis than RBC from individuals without diabetes (HbA1c <5.5 %) (See Fig. 2.4) (Qin et al. 2004). This reduced activity of CD59 in RBC from individuals with diabetes is consistent with inactivation of CD59 by glycation and would explain some reversible hematological abnormalities seen in individuals with poorly controlled diabetes including increased reticulocyte count and shorter RBC lifespan (Peterson et al. 1977; Bern et al. 1985; Giugliano 1982), both suggestive of mild hemolytic anemia. Also, individuals with diabetes experience a high incidence of thrombotic episodes, their platelets are hyperactive and their plasma contains high levels of platelet-derived microparticles (PMPs) (Carr 2001). Thrombosis is also a major cause of morbidity and mortality in individuals with PNH (Halperin and Nicholson-Weller 1989; Gardner et al. 1958; Hill et al. 2013) whose CD59-deficient platelets are exquisitely sensitive to MAC-induced activation and release of PMP (Wiedmer et al. 1993); platelets from CD59 knockout mice are also activated spontaneously and release PMPs (Qin et al. 2003, 2009a, b). These combined clinical and experimental observations indicate that inactivation of CD59 by glycation could be one factor contributing to the high incidence of thrombosis in individuals with diabetes.
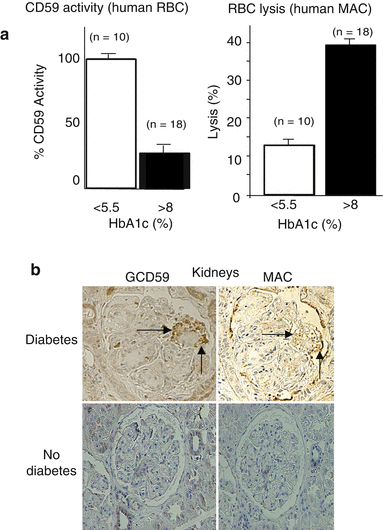
Fig. 2.4
MAC-mediated lysis (a) Reduced activity of CD59 and increased sensitivity to MAC-mediated lysis in RBC from individuals with diabetes. Red blood cells (RBCs) from individuals with diabetes (selected by HbA1c >8 %) have an 8–10 times lower CD59 activity than RBCs from individuals without diabetes (left panel), and are much more sensitive to cell lysis induced by addition of purified terminal complement components to form the MAC (right panel). (b) Co-localization of MAC and GCD59 in renal biopsies from individuals with diabetes. GCD59 co-localized with MAC in diabetic human kidneys. Serial sections of the same glomerulus in a kidney from an individual with and another without diabetes stained with anti-MAC or anti–GCD59 antibodies. Arrows indicate positive staining areas for glycated CD59 and MAC (For details on results depicted in this figure see Qin et al. (2004))
Diabetic patients exhibited glycated CD59 in organs with complications. An antibody that specifically recognizes the glycated from of CD59 (GCD59) was able to identify GCD59 colocalized with MAC in target organs of diabetic complications. The presence of GCD59 was confirmed in renal and sural nerve biopsies from individuals with diabetes. No detectable amounts of GCD59 were found in biopsies from individuals without diabetes (See Fig. 2.4b) (Qin et al. 2004). As expected from the functional inactivation of CD59 by glycation, serial sections of the same renal and nerve biopsies showed co-localization of GCD59 with MAC deposits (Qin et al. 2004). Increased levels GCD59 and MAC have also been observed in other tissues extracted from individuals with diabetes, namely skin biopsies and saphenous veins removed for grafting of occluded arteries (unpublished observation (Halperin)).
2.5 Complement-Targeted Therapeutics
Progressively recognized as a major mediator of a variety of clinical disorders that include both acute and chronic diseases and affect a wide range of organs, modulation of the complement system with either small molecules or biologics is now considered a promising strategy in drug discovery (Ricklin and Lambris 2007, 2013). Fueled by (a) a wealth of high resolution protein structures (Forneris et al. 2010; Janssen et al. 2005, 2006; Torreira et al. 2009; Wiesmann et al. 2006; Wu et al. 2009b), functional assays (Rooijakkers et al. 2009; Garlatti et al. 2010; Laursen et al. 2010; Shaw et al. 2010; Gingras et al. 2011; Kajander et al. 2011; Morgan et al. 2011, van den van den Elsen and Isenman 2011; Hadders and Gros 2012) and genome-wide association studies (Degn et al. 2011), which combined offer new insight into the molecular details and ligand interaction sites of complement components; (b) the approval and therapeutic success of the anti-C5 antibody, eculizumab, (Alexion, Cheshire, CT, USA) for the treatment of PNH (Hillmen et al. 2006, 2007; Rother et al. 2007) and more recently of atypical hemolytic–uremic syndrome (Legendre et al. 2013); and (3) the evidence that even small changes in the activity of individual complement proteins may add up to significantly affect disease progression, the field of complement-targeted drug discovery has seen a surge in approaches to therapeutically intervene at various levels of the complement cascades. Potential therapeutic approaches for interventions include inhibition of C3 activation, inhibition of C3 convertase assembly, acceleration of C3 convertase decay, promotion of C3B proteolysis, inhibition of C3 and/or C5 convertase activities, inhibition of membrane attack complex assembly by CD59, and reestablishment of normal alternative complement pathway control with protective Factor H protein. Compounds directed against some of these targets that are currently in early clinical trials for other indications include: compstatin/POT-4, a peptide inhibitor that inhibits the central step of the complement cascade by preventing cleavage of C3and ARC1905, a pegylated, aptamer-based C5 inhibitor that inhibits the cleavage of C5 into C5a and C5b. Other complement pathway-modulating compounds currently being considered for and/or are under preclinical development include a humanized anti-factor D antibody (TNX-234), a Fab fragment of an anti-complement factor B (CFB) antibody (TA106), a complement receptor 2 (CR2)-factor H hybrid proteins, a small molecule C5aR peptidomimetic (JPE-1375), and anti-properdin antibody, an inhibitor of C1 esterase inhibitor (C1-INH) and consequently the classical pathway, and a soluble form of complement receptor 1 (sCR1).
Based on the extensive clinical and experimental evidence supporting a role of complement in the pathogenesis of diabetes complications, it is conceivable that in the future, complement inhibition could offer an early point of intervention that could potentially retard, prevent, or even reverse progression of those complications. However, given the pleomorphic biological activities of the complement system, risk-benefit analyses of therapeutic application targeting complement to prevent or treat complications of diabetes will have to evaluate cautiously the potential systemic effects of chronic complement pathway modulation.
2.6 Glycated CD59 as a Biomarker for Human Diabetes
2.6.1 Available Biomarkers to Assess Glucose Physiology
The diagnosis of diabetes, appropriate medical management of diabetes, and risk stratification of diabetic complications all require reliable biomarkers that reflect integrated glucose handling. The most intuitive potential biomarker is circulating glucose itself; however, circulating glucose concentrations vary significantly based on the temporal relation to meals, physical activity, and stress. Therefore, standard practices include the measurement of a morning blood glucose while fasting, as well as more provocative physiologic maneuvers including measurement of glucose following a glucose challenge (such as the 75 g oral glucose tolerance test). A fasting morning blood glucose measurement provides cross-sectional circulating glucose concentrations independent of a prandial challenge, while the oral glucose tolerance test provides insight into the cross-sectional ability to handle a prandial glucose load. While both of these biomarkers of glucose handling are valuable, neither assesses long-term or integrated glucose handling over time.
Related posts:
 Cardiovascular and Metabolic Syndrome Impact on Uristatin
Cardiovascular and Metabolic Syndrome Impact on Uristatin
 C-Terminal Fragment of Adiponectin Receptor Clinical Correlations
C-Terminal Fragment of Adiponectin Receptor Clinical Correlations
 Development of Adiponectin Receptor C-Terminal Fragment Bioassays
Development of Adiponectin Receptor C-Terminal Fragment Bioassays
 Cell and Biological Models for the C Terminal Fragment of Adiponectin Receptor
Cell and Biological Models for the C Terminal Fragment of Adiponectin Receptor
 Uristatin Immunoassay Usage in Glomerular Nephritis Assessment
Uristatin Immunoassay Usage in Glomerular Nephritis Assessment
 Impact of Inflammation and Innate Immunity Response in Obesity Mediated Diabetes
Impact of Inflammation and Innate Immunity Response in Obesity Mediated Diabetes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


