DEFINITIONS AND HISTORY
Complement comprises a set of proteins present in plasma and other biological fluids, and on cell membranes, that together play key homeostatic roles in combating infection and disposal of waste. Complement is a central pillar of innate immunity, a ready-to-go, fast-response system that efficiently targets pathogens and toxic waste.
The discovery of complement dates back to the late 19th century when a number of pioneering biologists, including Josef Fodor, George Nuttal, and Hans Buchner, were exploring the bactericidal activities of plasma and serum.
1 They showed that fresh serum contained an activity that could efficiently kill some types of bacteria, and that this bactericidal activity decayed as the serum aged ex vivo and was rapidly lost when the serum was exposed to heat. To describe this heat-labile activity, Buchner coined the enigmatic term “alexin,” from the Greek and roughly translated as “without a name.” Jules Bordet extended these serum bactericidal studies using cholera bacilli and showed that sera from choleraimmune individuals efficiently killed the organism, whereas sera from nonimmune individuals did not; heating the immune serum caused loss of this bactericidal effect, suggesting it was related to Buchner’s alexin.
2 Bordet then did a clever experiment: first incubating cholera bacilli with heat-treated immune serum (which alone did nothing), then adding fresh nonimmune serum; the bacilli were killed, demonstrating that killing of cholera bacilli required two serum components, a heat-stable component present only in immune serum which he termed “sensitiser,” and a heat-labile component present only in fresh serum, alexin. Around the same time, Paul Ehrlich was exploring how immune serum caused hemolysis of animal erythrocytes; he also found that these same two components were required. The heat-stable component present only in immune serum he termed “amboceptor” (and later immune body or antibody), while the heat labile alexin, he called “complement,” to indicate that it merely complemented the inherent hemolytic effect of amboceptor.
3Over the first few years of the 20th century, an intense debate continued regarding the nature of complement and its relationship to antibody, with Bordet and Ehrlich as the main protagonists. The “complement fixation test,” developed by Bordet and his coworkers around 1900 as a means of testing whether an individual possessed antibodies against a particular bacterium (ie, was immune), relied on the fact that complement was consumed when antibody bound its target and demonstrated conclusively that complement was a distinct activity in serum.
4 This test, the mainstay of immune diagnostics for a century, is still used for some pathogens today. The famous Glasgow physician and pathologist Sir Robert Muir, writing in 1906, described complement as “that labile substance of normal serum that is taken up by the combination of an antigen and its antibody,”
5 a definition it would be hard to improve on today.
Over the next 20 or so years, a number of scientists used the serum fractionation techniques that were state of the art at the time to investigate the “substance” called complement. Euglobulin precipitation (by dialysing serum against water) revealed that neither the re-dissolved euglobulin precipitate nor the dialyzed serum supernatant alone possessed complement hemolytic activity; however, when re-combined, complement activity was restored, demonstrating the need for at least two components, termed C′1 and C′2. The precipitable euglobulin C′1 component was inactivated by heating to 56°C, while the soluble C′2 component was heat-stable. By the mid-1920s, other manipulations of serum, including adsorption on yeast particles, incubation with ammonia or treatment with cobra venom had shown that there were at least four separable components necessary for complement activity, termed C′1, C′2, C′3, and C′4.
6The physicochemical nature of complement also attracted much interest and debate until 1941, when a landmark paper from Louis Pillemer and his colleagues showed clearly that each of the components C′1 to C′4 was protein in nature.
7 Recognition of the protein nature of the complement components fueled a frenzy of protein chemistry activity that, by the mid-1960s, had further refined the components C′1 to C′4, in particular demonstrating that C′3 actually contained six separate proteins; the nine complement component proteins were therefore termed C′1, C′2, C′4, C′3a, C′3b, C′3c, C′3d, C′3e, and C′3f. The euglobulin, C′1, was also shown to be a complex of three different proteins. In 1968, a Committee on Complement Nomenclature met under the auspices of the World Health Organization to simplify and standardize, resulting in the modern terminology, in order of reaction, C1, C4, C2, C3, C5, C6, C7, C8, and C9.
8Up to the 1950s, complement research was focussed on the antibody-dependent activity recognised by Bordet and Ehrlich. Then, in 1954, Pillemer made a startling discovery: serum contained a protein that he called properdin (from the latin,
perdere, which means to destroy) that could trigger complement attack on pathogens without the need for antibody.
9 This finding was hailed as a “magic bullet” against infection and was so newsworthy that it was featured in
Time magazine under the banner “Medicine: Death to Germs.”
10 Others strongly denied the existence of the extraordinary properdin system, accusing Pillemer of carelessness or worse; perhaps in part due to this criticism, Pillemer committed suicide in 1958, leaving others in the 1960s to confirm and extend his findings. In 1971, Hans Muller-Eberhard and Manfred Mayer independently provided definitive proof of the existence of the properdin system, which they called the alternate pathway (now alternative) to distinguish from the original antibody-dependent classical pathway of activation.
11,12 Identification of complement proteins unique to this pathway soon followed.
Up until the end of the 1950s, complement proteins were considered, with no good evidence, to be minor plasma components, present in trace amounts and hence difficult to work with. This misconception was laid bare in 1960 with the demonstration by Hans Muller-Eberhard and colleagues that the β1c-globulin band visible on serum electrophoretograms, was in fact C3, a major component accounting for 1% to 2% of total plasma proteins.
13 Other complement proteins were soon shown to be relatively abundant in plasma, opening the door to their purification and functional characterization.
The next major leap forward was the recognition of the enzymatic nature of complement. Irwin Lepow and colleagues in the late 1950s had demonstrated that C′1 was associated with enzymatic (esterase) activity. By the mid-1960s, they had achieved a remarkable understanding of this first step of complement activation
14; they showed that the enzyme comprised three distinct protein subunits (C′1q, C′1r, C′1s), that formation of the C′1 complex required calcium ions, that its substrates were C′4 and C′2 in that order, and that the enzyme was controlled by a plasma C′1 esterase inhibitor. It was soon recognized that a puzzling disease, hereditary angioedema, long associated with complement activation, was caused by a deficiency of this inhibitor,
15 launching the field of complement therapeutics.
Around the same time, Muller-Eberhard and colleagues showed that activation of C′3 was also enzymatic in nature. They showed that a cell-bound complex of C′1-activated C′4 and C′2 could cause activation of many molecules of C3 and their deposition on the cell surface in an active form.
16 It took another 15 years before the mechanism by which activated C′4 and C′3 attached to membranes was identified, when Brian Tack’s group showed that both these molecules possess a labile, buried thioester group, exposed on activation, that covalently attached the proteins to surfaces.
17,18One problem with starting a history is knowing when to stop. The brief history above is incomplete and ends 40 years ago, and much that is noteworthy has occurred since then! However, that more recent history will form part of later descriptions, so I will draw a line here. Before moving on, I will remark that the use of “apostrophes” in complement nomenclature was quietly dropped in the late 1970s, so henceforth they will not be featured.
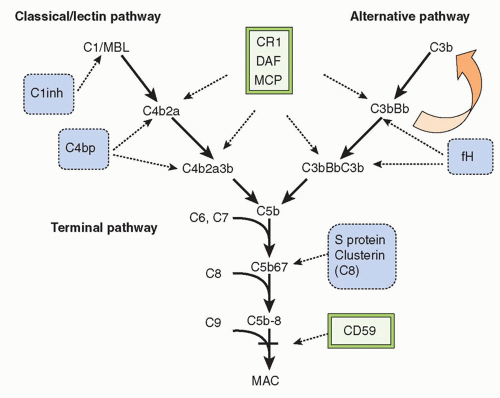
THE COMPONENTS AND PATHWAYS OF COMPLEMENT
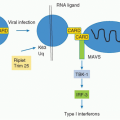
In the 21st century, the term
complement encompasses some 35 plasma and membrane proteins. The pathway components
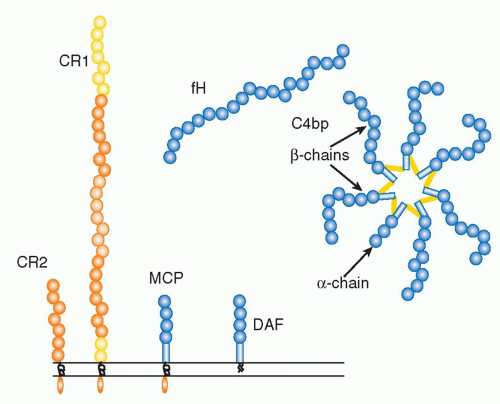
(Table 36.1) are outnumbered by regulatory proteins
(Table 36.2) that limit activation in plasma and on self cells, and receptors
(Table 36.3) that bind complement proteins to trigger a range of cellular responses. The complement proteins interact with one another to provide an effective and efficient antimicrobial defense system, along with a growing list of other roles, for example, in immune complex handling and priming for adaptive immune responses. Critical features include activation by diverse triggers, enzymatic amplification at multiple steps, and rigid control to prevent damage to self. Complement activation can be initiated in a variety of ways to target different pathogens and toxic agents. The literature describes three distinct activation pathways, although in reality, these pathways are closely interlinked. Antibodydependent activation, described by Ehrlich and others at the beginning of the 20th century, was for many years the only known pathway, now termed
the classical pathway (CP). The re-discovery by Pillemer in the 1950s of an antibodyindependent, pathogen-triggered pathway for complement activation laid the foundations for the
alternative pathway (
AP). A third activation pathway, antibody independent and triggered by pathogen-specific sugars, was described in the 1980s; at first given a variety of names to reflect the activating sugars, it is now universally known as the
lectin pathway (
LP). These three activation systems share common components but also have pathway-specific ones.
The Classical Pathway
Antibody-dependent triggering of the CP begins with immunoglobulin (Ig)G or IgM antibody bound to its target antigen either on a pathogen or host cell membrane, or on an immune complex. IgM, a large, pentameric molecule, is the most efficient activator of complement; a single IgM molecule bound to antigen is, in theory, sufficient to generate a nidus for complement activation. Free IgM in plasma does not activate complement; however, binding to antigen through its five antigen binding domains generates major structural change affecting the whole molecule, causing it to transition from a planar to a staple conformation. These events expose complement binding sites in the constant (Fc) regions of each of the five subunits that initiate the process of complement activation. In contrast, multiple IgG molecules, bound close together on the target, termed an immune array, are needed to trigger complement activation. Again, conformational changes occur, exposing complement initiation sites in the Fc regions of the molecule. Not all IgG subclasses possess complement binding sites; IgG1 and IgG3 are strong complement activators, whereas IgG2 is a weak activator and IgG4 does not activate complement at all.
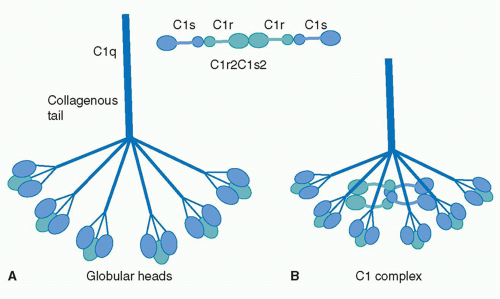
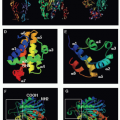
The first component of the CP is C1, a large multimeric protein comprising one copy of C1q and two copies each of C1r and C1s, held together noncovalently in a calcium-dependent complex. C1q, the recognition unit of the C1 complex, is itself multimeric, made up of six subunits, each comprising a collagenous stalk and a carboxy-terminal globular head; the six subunits are tightly associated along the collagenous stalks but separate in the head regions, giving the classical electron microscopy image of a “bunch
of tulips”
(Fig. 36.1). To add further complexity to this, the most complex of complement proteins, each of the six C1q subunits is itself assembled from a trimer of homologous chains termed C1q-A, C1q-B, and C1q-C, intertwined in a triple helix in the stalk but distinct in the globular head. C1r and C1s, the proteolytic units of the C1 complex, are homologous serine proteases that associate with one-another in a calcium-dependent linear tetramer complex in the order C1s-C1r-C1r-C1s. When bound in the C1 complex, the tetramer folds upon itself in a figure-eight conformation and sits between the globular heads, held in place by ionic bonds between acidic residues in C1r/C1s and a single basic residue in each of the six C1q stalks (see
Fig. 36.1).
19For C1 activation to occur, at least two of the six head domains must be engaged simultaneously, explaining both the efficiency of activation by the multivalent IgM and the
need for a critical surface density of IgG, the immune array, for activation; this latter limitation reduces the risk of inappropriate activation of complement on host tissues. Binding of the C1q globular heads to antibody Fc is itself a complex series of events; an exposed calcium ion in the head mediates the initial binding and induces further binding events that cause rotation of the head domain. These conformational changes stress the C1s-C1r-C1r-C1s tetramer tightly gripped between the C1q stalks, thereby triggering the autoactivation of C1r via cleavage at a single site to yield a two-chain disulphide-bonded active protease. Activated C1r then cleaves its homologous substrate, C1s, at a single site to generate the active C1s protease.
*,
19The C1s serine protease has two substrates, C4 and C2, the next two proteins in the classical pathway sequence. The illogical ordering here reflects the fact that complement components were named chronologically, according to the order of their discovery (C′1 > C′2 > C′3 > C′4), rather than according to their position in the reaction.
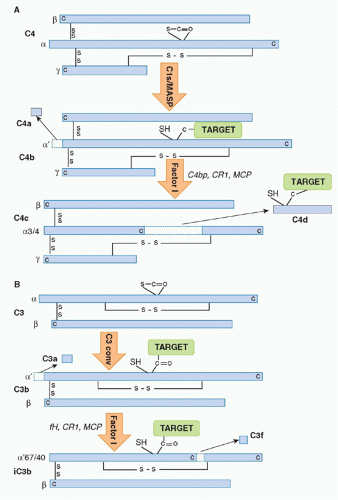
C4 is a relatively abundant protein, present in plasma at around 0.5 g/l. It is a large (210 kDa), disulphide-bonded heterotrimer. Activated C1s captures C4 from the fluid phase, perhaps in part through interaction of its short consensus repeat (SCR) domains with C4, then cleaves the C4 α chain at a single site near the amino terminus, releasing a 77 amino-acid fragment,
C4a, and exposing in the cleaved α′ chain of the large fragment
C4b a labile thioester group
(Fig. 36.2A). Although most of the nascent C4b formed will decay in the fluid phase through hydrolysis of the thioester, a small proportion will bind reactive hydroxyl or amino groups on the activating surface, creating a cluster of covalently bound C4b around the initiating IgG/C1 complex. Immobilized C4b binds the next component in the sequence,
C2, in a magnesium-dependent complex. C2 is a single-chain plasma protein of mass 100 kDa and plasma concentration around 25 mg/L; it is the most heat-labile of the complement proteins, destroyed by brief incubation of plasma at 56°C. C4b-bound C2 is cleaved by activated C1s in an adjacent IgG/C1 complex, releasing a 30 kDa fragment
C2b, while the 70 kDa
C2a fragment, an active serine protease, remains associated with C4b on the surface.
†,
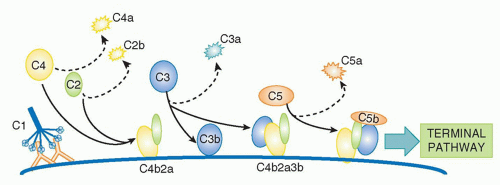
20,21The magnesium-dependent
C4b2a complex is the CP C3 convertase, the next activation enzyme in the sequence. C2a in the C4b2a complex is an active serine protease that cleaves
C3, a two-chain 190 kDa protein, homologous to C4 and the most abundant of the complement proteins at around 1 g/l in plasma.
22 Cleavage releases a 77 amino-acid fragment,
C3a, from the amino terminus of the α chain of C3, exposing in the large fragment,
C3b, a labile thioester group essentially as described previously for C4b
(Fig. 36.2B). Again, most of the C3b formed decays by thioester hydrolysis, but a small fraction covalently binds the activating surface, clustering around the site of activation. Some of the C3b formed will directly bind C4b2a through its thioester to form a trimolecular complex,
C4b2a3b; this binding is not a random event but occurs at a single specific site in C4b, placing C3b in the correct orientation for succeeding steps of activation. The C4b2a3b complex contains a binding site for
C5 involving interactions with both C4b and C3b in the complex. C5, another homologue of C4 and C3, is a 200 kDa, two-chain molecule present in plasma at about 100 mg/L; importantly, C5 lacks the critical thioester group and so cannot bind covalently to targets. Once bound to C4b2a3b, the CP C5 convertase, C5 is cleaved by C2a in the complex, releasing a 74 amino-acid fragment,
C5a, from the α chain of C5 and leaving the large fragment,
C5b, loosely attached to the convertase. Cleavage of C5 is the final enzymatic step in the CP
(Fig. 36.3).
Two features of CP activation are critical to its roles. First, amplification at each of the enzymatic steps is critical for efficient activation; thus, a single active IgG/C1 complex will deposit an abundance of C4b in the vicinity of the initiating
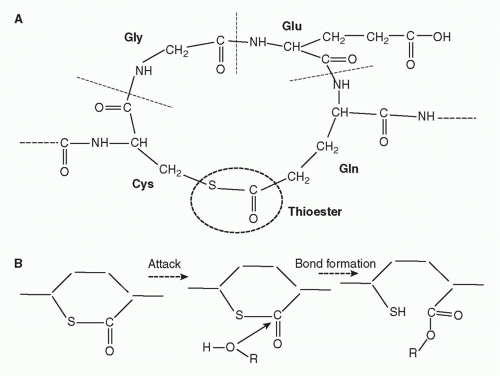
IgG/C1, each C4b2a complex formed will in turn cause deposition of many copies of C3b on the surrounding membrane, and each C5 convertase will also cleave multiple C5 molecules. Second, the nascent thioester groups in C4 and C3 are critical components of complement activation; without these entities, complement activation on surfaces would be an impossibility and the system would not function. The thioester group is formed from interaction between a glutamine and a cysteine residue that, in the intact molecule, are buried in the protein structure
(Fig. 36.4).
17,18 When activated by the convertase, a major conformational change occurs that exposes the internal thioester bond in C3b and C4b, making it very unstable and highly susceptible to attack by nucleophiles such as hydroxyl groups (-OH) and amine groups (-NH
2) in membrane proteins and carbohydrates, creating a covalent bond that locks C3b and C4b onto the surface.
23 The exposed thioester is highly labile and rapidly inactivated by hydrolysis, restricting binding of C3b and C4b to the immediate vicinity of the activating enzyme.
The Alternative Pathway
The concept of an AP of complement activation grew out of the recognition that some pathogens activated complement without the need for antibody. The controversial history was briefly described previously, but today the existence of the AP is not in doubt, although whether it should be considered a separate pathway remains an area of debate. The AP functions in two interlinked ways, first as an efficient amplification loop to drive further complement activation whatever the initiating trigger, and second as an always-on pathogen sensor ready to attack foreign surfaces.
24,25 The trigger for AP activation is the presence of “activated” C3; this can be “classically activated” nascent C3b on membranes or in plasma, or C3 that has been hydrolyzed in plasma,
C3(H2O). Spontaneous hydrolysis of C3 occurs under physiological conditions at approximately 5% of total C3 per day, driving a constant, low-grade “tickover” activation of the AP in plasma.
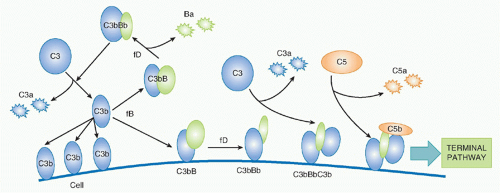
Factor B (fB), a 110 kDa single-chain protein and structural homologue of C2, binds to form a magnesium-dependent complex, C3bB or C3(H
2O) B; in its bound state, fB is cleaved by a plasma serine protease,
factor D (fD), releasing the 50kDa Ba fragment and leaving the serine protease fragment, Bb, in the complex to form the AP C3 convertase. The enzyme fD is a small (25 kDa) serine protease present in tiny amounts (1 mg/L) in plasma as an active enzyme with just one substrate, fB in complex with C3b/C3(H
2O). Bb in the C3bBb or C3(H
2O)Bb convertase cleaves C3 exactly as occurs in the CP, releasing C3a and generating C3b that can bind to targets and/or bind more fB to continue the AP amplification cycle—a positive feedback amplification loop
(Fig. 36.5). C3bBb on targets will catalyze the deposition of many C3b molecules as described for the CP convertase, and C3b binding to a specific site on C3b in the convertase will create a trimolecular C5 convertase in which C5 can be cleaved by the adjacent C2b, events that are analogous to those described in the CP. I have yet to mention the protein for which the AP was first named:
properdin. This complicated molecule, an oligomer comprising two, three, or four copies of a 53Da single-chain protein, binds and stabilizes the AP convertases, reducing their inherent tendency to fall apart (“decay”) and increasing markedly their capacity to perpetuate activation. Recently, a second role for properdin, binding to bacterial surfaces and forming a platform for AP convertase assembly, has been emphasised.
26The recognition that the AP is always on, in a constant tick-over state, explained how complement is so efficiently activated on pathogens. Tick-over ensures that all plasmaexposed surfaces are continuously showered with C3b; on self cells, inhibitory mechanisms described in the following sections ensure that no amplification occurs. However, on pathogens, lacking such protection, AP amplification kicks in, rapidly coating the surface with C3b. It is important
to note that the AP is inexorably linked to the CP in that C3b generated through the latter will feed into the former to amplify activation. It therefore does not matter whether the initial C3b is generated by the CP or AP (or, indeed, the LP that follows), the AP amplification loop will amplify the response, particularly when occurring on the intrinsically activating surfaces of pathogens.
The Lectin Pathway
The LP, first described only in 1987,
27,28 shares features with both the AP and CP. Like the AP, it provides antibody-independent “innate” immunity, activated by pathogens independent of antibody. Its similarities to the CP are legion; indeed, it only differs in the initiation step and is perhaps better considered as a different route to CP activation that bypasses the need for antibody.
29 The C1 complex is replaced by a structurally similar multimolecular complex, comprising a C1q-like recognition unit, either
mannan-binding lectin (MBL) or
ficolin (a family of three proteins in man), and
MBL-associated serine protease-2 (MASP-2) that provides the enzymatic activity. The recognition units bind carbohydrate epitopes, N-acetyl glucosamine for both ficolins and MBL, and mannose for MBL alone; these ligands are abundant in the cell walls of diverse pathogens, including bacteria, yeast, fungi, and viruses, making them targets for LP activation. Each MBL subunit comprises a homotrimer of 32 kDa chains, an amino-terminal collagen-like region responsible for trimerization in a triple helix, a short α-helical neck region, and a globular carbohydrate recognition domain. Subunits assemble into oligomers containing between two and six oligomers, the latter closely resembling the C1q hexamers. Indeed, C1q and MBL are members of the collectin family of proteins characterized by globular head regions with binding activities and long collagenous tail regions with diverse roles. Serum levels of MBL are highly variable in the population, from undetectable to 5 mg/L.
Ficolins are novel lectins, structurally similar to C1q and MBL through the collagenous regions, but with head regions comprising fibrinogen-like domains.
30 Three ficolins are described in man, termed ficolin-1, ficolin-2, and ficolin-3.
‡ Serum concentrations of all are low, ficolins-2 and -3 around 5 mg/L and ficolin-1 about 0.05 mg/L, although reported levels for each vary widely.
MASP-2 is a member of a family of homologous lectin-binding proteins, structural homologues of C1r/C1s, three of which express protease activity (MASP-1, MASP-2, MASP-3) while the other two do not (MAp19, also called sMAP; MAp44, also called MAP-1). After a decade of debate, claim and counterclaim, it is now generally agreed that MASP-2 is the critical enzyme of the lectin pathway: a MASP-2 dimer associates with the MBL or ficolin oligomer in a calcium-dependent manner to generate a complex that is necessary and sufficient to create the activation enzyme.
31 The biological roles of the other members of the MASP protein family remain obscure, although MASP-1 has recently been shown to be critical for AP activation in the mouse as it activates profD.
32 MASP-2 complexed with the MBL or ficolin oligomer captures C4 from the fluid phase, likely via its SCR domains, then cleaves C4 at a single site, identical to that targeted by C1s, to release C4a and generate C4b that binds through its thioester to the surface adjacent the initiating enzyme. The rest of the sequence mirrors that of the CP: C2 is captured onto C4b and presented for cleavage by MASP-2
in an adjacent complex, and the resultant enzyme, C4b2a, continues activation through C3 and C5.
The Terminal Pathway
The terminal pathway (TP), sometimes referred to as the membrane attack pathway, is a final, common pathway for all activation routes. Cleavage of C5 is the last enzymatic step in the complement sequence and the final step of each activation pathway. The TP is a system almost unique in nature where five plasma proteins join to create an amphipathic membrane-inserted complex, the membrane attack complex (MAC), that creates a lytic pore in the membrane
(Fig. 36.6).
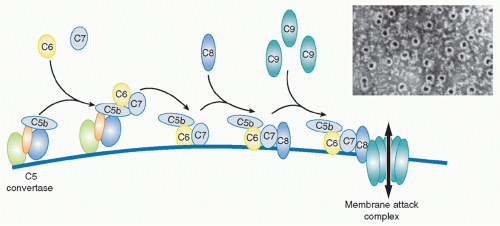
33 The TP begins with the binding of the next component in the sequence,
C6, to C5b still in the grip of the C5 convertase. C6 is a 100 kDa single-chain protein present in plasma at around 50 mg/L. Conformational changes during formation of the C5b6 complex weaken the grip of the convertase and create a binding site for the next component, C7, a 95 kDa single-chain molecule, plasma concentration about 90 mg/L. C6 and C7 are homologous molecules and are genetically linked, with genes adjacent on chromosome 5p. Incorporation of C7 causes further loosening of grip, releasing the trimolecular C5b67 complex into the fluid phase. The newly released C5b67 complexes shower down onto the lipid membrane surrounding the convertase and bind firmly to the surface via a hydrophobic site in the complex, thereby creating a nidus for continued assembly of the MAC. This is an inefficient process; the large majority of C5b67 complexes formed are inactivated in the fluid phase before they can bind membranes. Spontaneous inactivation occurs rapidly even when the C5b67 complex is assembled from pure proteins; in plasma, several proteins act as C5b-7 inhibitors to further accelerate inactivation. Those C5b67 complexes that do bind membranes then recruit the next protein in the sequence,
C8, a heterotrimeric molecule (α and β chains each approximately 61 kDa, γ chain, 22 kDa; α and γ covalently linked, β noncovalently associated) present in plasma at about 80 mg/L. Binding of C8 introduces additional hydrophobicity, causing the resultant C5b-8 complex to embed more firmly in the membrane. There is some evidence that the C5b-8 complex can cause membrane disruption and leakiness, but the major membrane disruption necessary to kill bacteria or other target cells requires the recruitment of multiple copies of the final component of the MAC,
C9, a 70 kDa single-chain protein present in plasma at around 60 mg/L. The first globular C9 molecule binds C8 in the C5b-8 complex and undergoes major conformational rearrangement, unfolding to reveal a hydrophobic face that allows insertion into and through the membrane lipid bilayer. As additional C9 molecules are recruited, they in turn unfold and insert, aligning with the first C9, like barrel staves; with the recruitment of about 10 C9 molecules, the barrel is completed, creating a protein-lined channel through the membrane, the MAC (see
Fig. 36.6). The C9 hydrophobic faces tightly lock the MAC in the membrane while the opposite, hydrophilic faces create a channel through which water and ions can flow, the MAC pore.
** In electron micrographs of complement-lyzed targets, the MAC is readily visible as a
ring of about 10 nm internal diameter. The C5b-8 complex is displaced to the edge of the ring, resembling a pan handle.