The treatment of colorectal cancer (CRC) is evolving continuously and many important advances have recently led to improved prognosis. Surgery remains an integral component of multidisciplinary treatment. Targeted therapies are being developed in the laboratory and implemented clinically, thus taking advantage of major advances in understanding the molecular biology of CRC. This chapter briefly reviews the epidemiology of CRC and then the molecular biology of CRC and discusses inherited CRC syndromes, and also CRC screening and surveillance.
Colorectal cancer is the third most common cancer in the United States in both men and women and the third most common cause of cancer-related death in the United States, accounting for more than 50,000 deaths annually.1 Worldwide, CRC affects over a million people and the incidence appears to be increasing globally.2 In the United States, the incidence of CRC is declining, possibly as the result of improved screening and early detection. However, the risk of developing CRC remains approximately 1 in 21 for women and 1 in 19 for men.1 The average age of diagnosis is 72 years.3 Age is a strong risk factor for CRC and the vast majority of cases occur in patients over 50 years of age.1,4,5 The probability of developing CRC for a patient less than 40 is less than 1 in 1000, but increases to approximately 1 in 25 for patients 70 years or older. Interestingly, though the incidence of CRC is decreasing for the U.S. population in general, it appears to be slightly increasing in younger patients.6 African Americans develop CRC with increased frequency compared to Caucasians, a fact which is relevant to newer screening guidelines.
In general survival for CRC is better compared to other GI malignancies. Five-year survival for patients presenting with all stages of CRC is 64%.1 Survival is clearly influenced by the stage of disease at presentation. Five-year survival for patients with localized CRC is approximately 90%, compared to 69% for regional disease and 12% for all patients with metastatic disease.7 This underscores the importance of early diagnosis. When appropriately diagnosed, CRC is a survivable disease. There are over 1 million CRC survivors alive in the United States.8
Polyps are localized projections of mucosa above the level of the surrounding tissue.5,9 Polyps can be broadly classified according to malignant potential. Benign polyps include hyperplastic, hamartomatous, and inflammatory. Polyps with neoplastic potential include adenomatous, serrated, and hamartomatous polyps (Table 106-1). Adenomatous polyps are the most common, representing about 65% to 75% of identified polyps.5,10 These polyps can be further classified based on histology as tubular, tubulovillous, or villous.
Classification of Polypsa
| Histologic Classification | Features | Molecular Features |
|---|---|---|
| Adenoma | Most common, dysplasia present. Malignant potential | Classic adenoma to carcinoma pathway with mutation in APC, KRAS, and CIMP rare |
| Hyperplastic polyps (HP) | Typically are located in distal colon | |
| Goblet cell-rich type (GCHP) | Abundance of goblet cells | KRAS mutation common |
| Microvesicular type (MVHP) | Mucin filled vesicles | BRAF mutation common |
| Sessile serrated adenoma (SSA) | Some degree of cellular atypia such as architectural distortion, but not typically dysplasia | BRAF mutation common (75%–82%) and CIMP high is common (92%) |
| Traditional serrated adenoma (TSA) | Rare entity characterized by adenomatous dysplasia | Heterogenous |
| Mixed serrated polyp (MP) | Term used when a SSA develops evidence of dysplasia | BRAF commonly mutated |
Serrated polyps are characterized by a “saw-toothed” appearance on histology and represent another class of polyps that has recently gained attention as an important contributing pathway to CRC.11,12 The nomenclature for hyperplastic polyps is somewhat inconsistent, and therefore confusing. One classification is represented in Table 106-1.12 This group includes benign hyperplastic polyps as well as the sessile serrated adenoma (SSA) and the traditional serrated adenoma (TSA).11,12 SSAs are important, as they occur more commonly in the proximal colon compared to adenomatous polyps10 and the benign hyperplastic polyp.13 As discussed in more detail below, SSAs have fairly recently been shown to harbor malignant potential and represent a new pathway for the development of CRC.12,14,15
Hamartomatous polyps demonstrate marked architectural abnormality without evidence of dysplasia.9,16 These polyps are characteristic of the hamartomatous polyposis syndromes which include juvenile polyposis syndrome, Peutz–Jeghers syndrome, and the PTEN hamartoma tumor syndrome.16
Muto first described clinically the adenoma to carcinoma sequence in 1976.17 Fearon and Vogelstein proposed the molecular pathways in the adenoma to carcinoma sequence.18 Many small (i.e., <5 mm) polyps are hyperplastic and are not thought to be precursor lesions to the majority of cancers.5 Adenomas occur when polyps display abnormal morphology and differentiation within the lesion. By definition, adenomatous polyps have low-grade dysplasia. The Vogelstein concept is that stepwise, sequential, and genetic derangements occur within colonic mucosa. As mutations continue to accumulate, an adenoma will eventually develop into an invasive cancer.5
This concept gained widespread support due to several observations. Polyps are common and their frequency increases with age, as does the incidence of CRC.19 Frequently carcinoma is found in association with adenomatous polyps.5 Also, removal of polyps lowers the risk of subsequent development of CRC. Data for this is provided by the National Polyp Study, a multicenter study which demonstrated that colonoscopic polypectomy reduced mortality from CRC.20 A recent update of this study demonstrated that polypectomy was associated with a 53% reduction in deaths from CRC.21
The malignant potential of a polyp is related to its size, histology, and grade of dysplasia.17,22 Larger polyps are more likely to result in subsequent invasive cancer; a 1-cm tubular adenoma carries approximately 5% cancer risk compared to a 2-cm tubular adenoma, which carries a risk as high as 35%.22 A villous adenoma measuring 2 cm carries approximately a 50% risk. Finally, one can see the influence of polyps in patients with familial adenomatous polyposis (FAP) syndrome, who develop hundreds of polyps, and have a near 100% rate of developing malignancy (discussed in more detail below).
Identification of the importance of the SSA has led to a broader concept of CRC progression via multiple polyp pathways that likely represent variable genetic derangements. Hyperplastic polyps were initially thought to be benign and not contributive to the development of CRC. However, in the setting of hyperplastic polyposis syndrome, the malignant potential of serrated polyps was recognized,12 and it is now recognized that hyperplastic polyps exist within a spectrum and that SSAs do harbor malignant potential.11,12
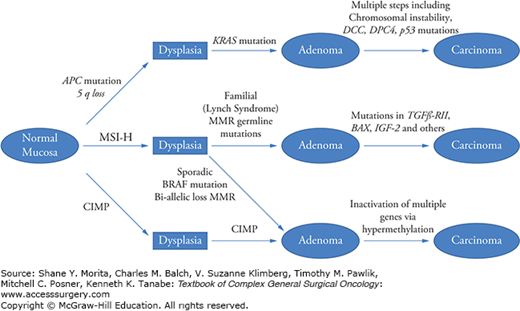
The broad classifications of adenomatous and serrated lesions allows for consideration of the development of CRC via several molecular pathways, as shown in Fig. 106-1. Adenomatous polyps develop along the more traditional pathway involving APC mutation, KRas mutation, and chromosomal instability. This pathway accounts for approximately 75% to 80% of CRCs. The remaining 20% to 25% of patients develop CRC via a separate pathway involving mechanisms of microsatellite instability (MSI) and CpG island promoter hypermethylation. Serrated polyps most commonly develop BRAF mutations, though a smaller proportion will demonstrate KRAS mutations. In approximately 15% of sporadic CRC, MSI develops due to hypermethylation of MLH1, which leads to a CpG island methylator phenotype-high (CIMP-H) tumor, also known as a mutator phenyotype. Germline mutation of one allele of a mismatch repair (MMR) gene occurs in Lynch syndrome and accounts for 2% to 3% of CRC. This is discussed in more detail below.
FIGURE 106-1
Pathways of CRC development. There are several molecular pathways for the development of CRC. Most cancers arise from the classic pathway in which an adenomatous polyp forms and subsequent chromosomal instability arises. However, MSI-H tumors develop either through adenomas in the case of inherited lesions (HNPCC) or serrated lesions in the case of sporadic cancer. Sporadic MSI-H tumors are typically BRAF mutant. CpG island methylator phenotype (CIMP) tumors develop via serrated lesions and promoter hypermethylation is the driver for suppression of tumor suppressor genes. (Adapted with permission from Fearon ER. Molecular genetics of colorectal cancer.Annu Rev Pathol. 2011;6:479–507.)
All cancers, including CRC, are thought to be related to genetic changes resulting in aberrant protein expression. The molecular characteristics of CRC are becoming increasingly important for diagnosis and treatment of patients with CRC. A full understanding of the molecular biology of CRC is critical to the treatment of the disease, as patients are increasingly likely to receive targeted treatments that will depend on the molecular characteristics of their tumor.
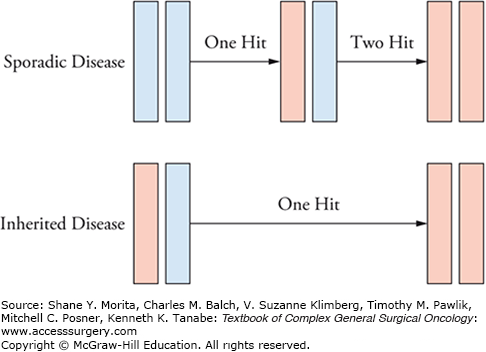
Mutations can occur in either proto-oncogenes or in tumor suppressor genes (TSGs). The term proto-oncogene refers to the normal gene before mutations occur. Oncogenes are mutated genes which result in a gain-of-function mutation. TSGs are those that are involved in checkpoint regulation. When TSGs are mutated, loss of function may result in uncontrolled cell growth which when combined with other genetic changes can lead to cancer. This requires a dual mutation, as proposed in Knudson’s two-hit hypothesis.23 Most hereditary cancer syndromes involve germline mutation of one allele of a TSG. The second allele is lost at a later time in somatic cells (Fig. 106-2). Several key pathways involved in the development of CRC have been identified. The following section outlines several of the important molecular pathways in CRC.
FIGURE 106-2
Knudson’s two-hit hypothesis. In sporadic disease, a single “hit” results in a heterozygous state which will not result in disease if the mutation is in a tumor suppressor gene. However, in inherited disease, a germline mutation of a TSG makes subsequent loss of function more likely as only one hit is required.
Genetic instability can occur through direct DNA damage, such as that done by ultraviolet light or by metabolic byproducts. Replication errors constitute another source of DNA errors. DNA replication occurs in preparation for mitosis. During this process, the two parental DNA strands open and serve as a template strand for the replication of a daughter strand. Errors that occur during replication must be repaired to prevent the accumulation of genetic defects. Several mechanisms exist to perform such repair, and mutations in these pathways predispose to various cancers.24,25 One such pathway is the mismatch repair pathway.
DNA contains regions of short tandem repeats of mono- or dinucleotide base pairs. These are most commonly located in noncoding regions of the DNA, but may be found anywhere within the DNA molecule. The length of each microsatellite region is constant within a patient. When replication errors occur, the number of repeats changes. Intact MMR machinery allows for correction of this error. Mutations that affect MMR genes result in microsatellite instability.
Microsatellite regions tend to exhibit a higher rate of mismatches, thus making these regions particularly susceptible to defects in MMR genes. Six MMR genes are involved in the human MMR system, hMLH1, hMSH2, hMSH3, hMSH6, hPMS1, hPMS2. Germline mutations in hMLH1 and hMSH2 represent the majority of identified mutations in Lynch syndrome (see below for full description of Lynch syndrome). In tumors with defects in MMR genes, replicative errors in microsatellite regions are not repaired, which can lead to silencing of proto-oncogenes or TSGs that contain microsatellites. Examples of such genes are numerous, but include transforming growth factor receptor beta (TGFR-β), insulin-like growth factor (IGF), BAX, and PTEN.26 Tumors with such mutations in MMR can develop a hypermutable phenotype that allows for the accumulation of mutations at a faster rate than in microsatellite stable (MSS) tumors.26,27 This high rate of mutations may be responsible for those CRC that develop quickly following a normal colonoscopy.28
Microsatellite instability has been defined either by the number of altered microsatellites or the percentage of alterations in the microsatellites tested. A tumor is considered to demonstrate microsatellite instability (MSI-H) if greater than or equal to two of the five markers tested are altered or if >30% of the total microsatellites tested are altered (if greater than five tested).29,26 There are also monomorphic microsatellite markers such as BAT26 or BAT25 which, if altered, suggest that the tumor is MSI-H. These markers may be useful for screening purposes.30
MSI-H, or mismatch repair deficient tumors, exhibit properties that differentiate them from MSS, or mismatch proficient, tumors. Sporadic MSI-H tumors tend to occur in older females and are mostly right sided. Histologically they are more commonly high-grade, have mucinous features and demonstrate tumor infiltrating lymphocytes, and Crohn’s-like reaction. MSI-H tumors are typically associated with a better prognosis compared to MSS tumors of the same stage.29,31
Germline mutation in the APC gene results in the development of hundreds of polyps and is associated with FAP syndrome. However, somatic mutations in the APC gene occur in approximately 70% to 80% of sporadic CRC as well.27 APC is a 300-kDa protein that is highly expressed in the colon,32 though it is present in all organs. APC likely has multiple functions involved in cell adhesion, proliferation, and apoptosis. One of the major functions of APC involves the β-catenin-dependent Wnt pathway, known as the canonical Wnt pathway to distinguish it from β-catenin-independent Wnt signaling.5 Wnt signaling occurs in stem cells located at the base of colonic crypts. This signaling allows for the proliferation of new enterocytes and their proper migration away from the base of the crypt. In the presence of Wnt, β-catenin migrates to the nucleus and activates target genes responsible for migration and proliferation.33
Currently, it is suggested that APC forms part of a multiprotein assembly known as the destruction complex, which consists of APC and a scaffold component such as axin or conductin.33,34 The destruction complex binds β-catenin and targets the protein for ubiquitination and subsequent proteosomal degradation.5,34 In the situation in which APC is mutated and does not inhibit β-catenin, β-catenin accumulates in the cytoplasm and nucleus.34 This accumulation of free β-catenin mimics constitutive activation of the Wnt pathway. β-catenin subsequently translocates to the nucleus, resulting in activation of multiple transcription factors such as δPPAR.5,34 Approximately half of the colon cancers that do not exhibit APC mutation will exhibit mutation in β-catenin, demonstrating the importance of this pathway in the initiation of neoplasms.33,35
In addition, APC mutation allows for chromosomal instability to occur through loss of the function of APC during mitosis.34,36–38 APC is involved in stabilizing the chromosome during mitosis. Loss of this function leads to the development of chromosomal instability.34
The RAS family of proteins was initially identified as rat sarcoma factors, and RAS was the first oncogene identified.39,40 The key members of this family include KRAS, HRAS, and NRAS. These proteins function as GTP/GDP-binding signal transduction switches, relaying the signal from extracellular molecules to the nucleus. This cascade eventually results in downstream activation of mitogen-activated protein kinase (MAPK) or phosphatidylinositol 3-kinase (PI3K) pathways, and ultimately helps regulate cell proliferation and survival.5,39,40
RAS mutations are common in CRC; KRAS mutations occur in approximately 40% of patients.5,41 In the normal state, RAS is present in the GDP-bound form. Activation occurs when external stimuli result in binding of the receptor tyrosine kinase. This causes a transformation into the GTP-bound form, which activates downstream kinases. The process is limited by RAS-GTPase activating proteins (RAS-GAPs), which deactivate RAS-GTP back to the RAS-GDP state. The most common mutations of KRAS result in conformational changes which prevent RAS-GAPs from deactivating RAS-GTP; the result is constitutive activation and amplification of downstream pathways.
BRAF is downstream of RAS in the MAP kinase pathway. Mutations in BRAF occur in approximately 10% of patients with CRC. BRAF mutations occur more commonly in sporadic MSI high tumors, and the mutation is frequently seen in SSA.42 BRAF mutant tumors are resistant to certain targeted therapy.43–46 BRAF mutations are associated with right-sided lesions and mucinous or poorly differentiated histology.45,46 BRAF and KRAS mutations are thought to be mutually exclusive.47 Evaluation of polyps demonstrating mutation in BRAF and KRAS suggest that these mutations may be early occurrences in the development of carcinomas via the serrated adenoma pathway.42,47
Familial CRC represents approximately 20% of all CRC, including patients with well-defined syndromes as well as those who display a clear family predisposition but do not meet criteria for a defined syndrome.48–50 Hereditary CRC is an important entity, as proper identification allows for screening of potentially affected family members and intervention at an early opportunity. Table 106-2 provides an overview of common hereditary CRC syndromes.
Characteristics of Hereditary Cancer Syndromes (please refer to text for references)
| Syndrome | Genetics | Polyps | Age of CRC | Extracolonic Manifestations |
|---|---|---|---|---|
| Familial adenomatous polyposis (FAP) |
| 100s to 1000s | 39 | Fundic gland polyps, duodenal adenomas, and cancer; desmoid tumors; congenital hypertrophic retinal pigment epithelium (CHRPE); osteomas; thyroid cancer; brain malignancy |
| Attenuated familial adenomatous polyposis (AFAP) |
| Up to 100 | 55 | Similar upper GI manifestions to FAP, but desmoids, osteomas and CHRPE are less common |
| MutYH-associated polyposis (MAP) |
| Frequently mimics AFAP but can reach 1000s of polyps | Duodenal adenomas and others similar to FAP | |
| Lynch syndrome |
| Less than 100 | 45 | Endometrial cancer, gastric cancer, transitional cell carcinoma (TCC) of renal pelvis and ureter, small bowel cancer, sebaceous adenomas and others |
| Peutz–Jeghers syndrome | STK11/LKB1 | Yes | 40 | Hamartomatous polyps throughout GI tract; skin pigmentation of perineum, perioral, hands, feet, and genitalia; increased risk of breast, biliary, gallbladder, ovarian, and Sertoli cell tumors. |
| Juvenile polyposis | SMAD4 | Yes | 60 | Polyps throughout GI tract, congenital abnormalities |
| POLE and POLD1 associated polyposis (DNA proofreading-associated polyposis) (PPAP) | DNA polymerases | Less than 100 | 30–50 | POLE D1 endometrial cancer |
Familial adenomatous polyposis syndrome is, perhaps, the best example of a genetic predisposition to cancer. Reports of families with numerous polyps date back more than 100 years. The mutation in the APC gene was identified on chromosome 5q21.51 The classic syndrome describes cases of patients in which there is the development of hundreds to thousands of gastrointestinal polyps, most notably in the large bowel. Extraintestinal manifestations are also present and contribute significantly to the morbidity and mortality associated with FAP.
The syndrome is inherited in an autosomal dominant fashion with nearly 100% penetrance.48 Germline APC mutations occur in about 1:10,000 live births. About 20% to 25% of mutations develop without a family history, that is, de novo mutations.5,52 The majority of APC mutations are inactivating mutations which result in loss of a TSG. A number of studies have demonstrated that the location of APC mutation determines the phenotype variation in FAP patients (i.e., genotype–phenotype correlation). Nearly 100% of patients with FAP will develop cancer of the colon without treatment, at an average age of 39 years.48 Table 106-3 indicates many of the known genotype–phenotype correlations.
Genotype–Phenotype Correlations in FAP
| Phenotype | Genotype |
|---|---|
| Severe polyposis | Mutations between codons 1250 and 1454 |
| Desmoid tumors | Beyond 1444 |
| Attenuated polyposis | Mutations of 3′ and 5′ ends of APC gene Exon 9 |
| Rectal cancer risk | Mutations between codons1250 and 1464 |
| Osteomas | Mutations between 767 and 1513 |
| Congenital hypertrophic retinal pigment epithelium (CHRPE) | Exons 9–13 |
The principal manifestation of FAP is the development of colonic polyps. The phenotype that develops can be described based on the number of polyps identified. The classical manifestation is that of over 100 polyps.48 Those with less than 100 polyps have an attenuated form of FAP (AFAP). The latter patients harbor mutations in the 3′ and 5′ ends of the APC gene and in exon 9, whereas those with greater than 1000 polyps have a form considered to be profuse, and this is associated with mutations in the region between codons 1250 and 1454.53
Surgical management of the colon in patients with FAP includes a prophylactic colectomy. The options for resection include an abdominal colectomy with ileorectal anastomosis (IRA), restorative proctocolectomy with ileal pouch anal anastomosis (IPAA), or proctocolectomy with end ileostomy. The latter procedure is no longer commonly performed unless there is a rectal cancer and the sphincter cannot be saved, poor rectal tone, or lifestyle issues. The disadvantage of the IRA is that the remaining rectum is at risk for development of polyps and subsequent rectal cancer.48 An IPAA reduces the risk of rectal cancer, but carries a higher morbidity, including quality of life issues such as stool incontinence, night seepage, potential sexual and urinary dysfunction, and other deleterious symptoms. Nevertheless, IPAA is a good procedure, and is the favored procedure for FAP in some centers. Some suggest that mutation information may be helpful to guide treatment. Those with a mutation in the region between codons 1250 and 1464 and a severe phenotype are the most likely to develop profuse polyposis and rectal cancer, and would probably benefit from an upfront pouch.54,55 Better patient selection for IRA has led to decreased incidence of cancer in the remainder of the rectum, as more patients with severe polyposis are now undergoing an IPAA.56 In this era in which IPAA is more commonly used, those patients with few rectal polyps can undergo IRA, with an acceptable risk of rectal cancer.57 In addition to the size and number of polyps, the presence of cancer, age of the patient, family history of desmoid tumors, female fecundity, surgeon’s experience, and patient’s input are other factors that may influence the type of procedure performed. It should be noted that all patients need postoperative surveillance, as even those with IPAA can develop cancer.
Osteomas are benign bone growths that occur in approximately 20% of patients with FAP.58 Patients with mutations in codons 767 to 1513 are at risk for the development of these lesions.59 The term Gardner syndrome was previously used to describe patients who developed multiple colorectal adenomas, CRC, and osteomas. As the molecular genetics of APC mutation and FAP become better understood, it is now recognized that Gardner syndrome is a variant of FAP based on the genotype–phenotype relationship of the specific APC mutation, and this term is no longer widely used.32,49,59 Osteomas in general do not require treatment. At times they are excised for cosmetic purposes.
There is an association between colorectal polyposis syndromes and the development of brain tumors. Approximately 1% to 2% of patients with FAP develop brain tumors. The majority of these are medulloblastomas, but astrocytomas and ependymomas can occur.60 In patients with Lynch syndrome the primary tumor which develops is an astrocytoma or glioblastoma. Treatment for patients with FAP and medulloblastoma involves multidisciplinary care with radiation, surgery, and possibly chemotherapy.58,61,62
Desmoid tumors are another extraintestinal manifestation of FAP that occur in approximately 15% to 25% of affected patients.49,55,63,64 They are mesenchymal tumors composed of a benign proliferation of fibroblasts which demonstrate a wide spectrum of behavior. Desmoid tumors are rare outside of FAP; they are approximately 1000 times more common in FAP compared to the general population.49 Desmoids lack metastatic potential, but are locally invasive. They are frequently associated with the small bowel mesentery, and this contributes to their morbidity.63 In patients with FAP, intra-abdominal and abdominal wall desmoids are more common than extra-abdominal wall desmoids (in contrast to sporadic cases). Patients also frequently develop multiple tumors.49,63 There is a genetic predisposition to developing desmoids in patients with mutations beyond the 1444 codon.49
In order to better compare patients with desmoid tumors, a staging system has been developed to classify patients65,66 (Table 106-4). This system classifies patients into four stages based on size of tumor, symptoms, and rate of growth. Patients with stage IV desmoids have been shown to have a worse prognosis, with an increased risk of developing symptoms and dying from desmoid disease.66 Treatment of intra-abdominal desmoids is also guided by stage. Stage I tumors are small, and can be managed with observation or with nonsteroidal anti-inflammatory drugs (NSAIDs), typically sulindac.49,65 Stage II tumors are associated with mild symptoms, and are less than 10 cm and stable in size. These tumors can be managed with resection if possible. Otherwise, treatment with NSAIDs or tamoxifen is appropriate. Stage III tumors are 10 to 20 cm, are associated with moderate symptoms, or are slowly growing. These tumors are typically managed with antiestrogen therapy or NSAIDs initially, with chemotherapy given if these approaches do not work. Finally, stage IV tumors cause significant symptoms, are greater than 20 cm, or are rapidly growing. These tumors are frequently the cause of desmoid-related mortality. Treatment may require surgery due to complications such as bleeding or perforation. However, there is a high rate of associated complications, including short gut syndrome.65,66 Recurrence of desmoids is the rule rather than the exception, and surgical treatment should be undertaken only after extensive consideration, frequently as a last resort.
Related posts:
 Defining the Specialty of Surgical Oncology
Defining the Specialty of Surgical Oncology
 Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Multimodality Therapy of Gastric Cancer: Eastern Experience
Medullary Thyroid Cancer
Landmark Clinical Trials that Impacted Surgical Management of Invasive and Noninvasive Breast Cancer
Multimodality Therapy of Gastric Cancer: Eastern Experience
 Cystic Lesions of the Liver and Biliary Tract
Cystic Lesions of the Liver and Biliary Tract
 Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Skin Closure After Resection Of Skin Malignancies, Including Melanoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


