Colorectal cancer (CRC) is a common disease, and approximately 25% of patients have a familial component. High-penetrance singlegene germline mutations conferring a true hereditary susceptibility account for around 5% to 6% of all cases. Lynch syndrome is the most common hereditary form of colorectal cancer. Much of the hereditary component in the remaining familial cases of CRC is likely polygenic, and many of the genetic changes involved are as yet unidentified. This article addresses the most clinically important CRC genetic syndromes.
Colorectal cancer (CRC) is a significant worldwide health care problem. Global cancer statistics from 2002 report CRC as the third most common cancer with around a million cases and the third and fourth most common cause of cancer-related mortality in men and women, respectively, accounting for approximately 508,000 deaths. In the United States, in 2009 there were an estimated 149,000 new cases of CRC with 49,000 deaths (second most common). Most (70%–80%) CRCs are sporadic. Around 20% to 30% of cases have a familial component, that is, one or more affected first- or second-degree relatives (or both), and a potentially definable genetic basis ( Fig. 1 ). In the United States, around 10% to 15% of adults have a history of CRC in a first-degree relative. Because early diagnosis of CRC is directly related to prognosis, there has been much interest in screening for this disease. Compelling evidence supports screening in average-risk individuals older than 50 years to reduce CRC mortality by detecting cancer at an early stage and by detecting and removing clinically significant adenomas. Early detection is especially important in high-risk groups (those with a CRC-positive family history) because CRC in this setting tends to occur at an earlier age compared with the average-risk population and may result in a disproportionate loss of years of life.
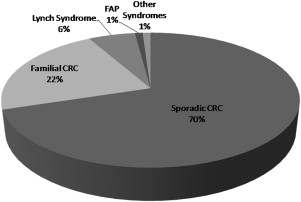
Over the last 10 to 15 years, molecular genetics has made a significant impact by identifying germline and somatic mutations associated with the development of CRC. Single-gene germline mutations conferring a hereditary susceptibility to CRC account for around 6% to 7% of all CRCs. Mutations in DNA repair genes ( MLH1 , MSH2 , MSH6 , PMS2 , MYH ) as well as in genes involved in signal transduction ( APC , SMAD4 ) are associated with true hereditary syndromes. Much of the inherited components in the remaining familial cases of CRC is likely polygenic, and many of the genetic changes involved are as yet unidentified. This article addresses the most important CRC genetic syndromes.
Lynch syndrome, DNA mismatch repair, and microsatellite instability
Lynch syndrome (LS; previously known as hereditary nonpolyposis CRC, HNPCC) is the most common form of hereditary CRC. In the early 1990s, linkage analysis discovered the genetic basis of LS with the identification of germline mutations in the DNA mismatch repair (MMR) genes MLH1 and MSH2 . Mutations in these genes explained more than 90% of LS cases. Subsequently, germline mutations in the MSH6 and PMS2 MMR genes were also shown to result in LS and may be more common than initially thought. Rare atypical LS pedigrees with identifiable MLH3 mutations have also been described. The phenotypic spectrum of LS includes cancers of the colorectum (right-sided colon in up to two-thirds of cases), endometrium, stomach, upper urinary tract, small bowel, ovary, bile ducts, brain, and skin (sebaceous tumors and keratoacanthomas in the Muir-Torre variant [frequently caused by MSH2 mutations]). Clinical criteria based on family history, the Amsterdam I and II criteria ( Table 1 ), were originally devised to identify patients with an inherited form of CRC, that is, HNPCC. However, problems were encountered with these criteria because these identified a heterogeneous group of patients with some cancers caused by MMR gene mutations and others caused by MMR gene inactivation or unknown etiology. Recently, it has been proposed to use the term LS to specifically refer to those patients with HNPCC who carry a pathogenic mutation in DNA MMR genes. An earlier-onset syndrome, known as constitutional MMR deficiency syndrome, occurs in the presence of homozygous or heterozygous mutations in MMR genes, especially PMS2 .
| Classic ICG-HNPCC (Amsterdam I) Criteria | Revised ICG-HNPCC (Amsterdam II) Criteria | Revised Bethesda Guidelines (for MSI Testing) |
|---|---|---|
| Three relatives (or more) with CRC (at least 1 of whom is a first-degree relative of the other 2) | Three relatives (or more) with an HNPCC-associated cancer a (at least 1 of whom is a first-degree relative of the other 2) | CRC diagnosed at age <50 y |
| Two successive generations (or more) with CRC | Two successive generations (or more) with an HNPCC-associated cancer | Presence of synchronous, metachronous colorectal, or other HNPCC-related tumors a regardless of age |
| One family member with CRC diagnosed before age 50 y | One family member with an HNPCC-associated cancer diagnosed before age 50 y | CRC diagnosed in 1 or more first-degree relatives with an HNPCC-related tumor with 1 of the cancers being diagnosed under age 50 y |
| FAP should be excluded | FAP should be excluded in the CRC cases, if any | CRC diagnosed in 2 or more first- or second-degree relatives with HNPCC-related tumors, regardless of age |
| Tumors should be verified by pathologic examination | Tumors should be verified by pathologic examination | CRC with MSI-H histology b diagnosed in a patient who is younger than 60 y |
a Colorectal, endometrial, stomach, ovarian, pancreas, ureter and renal pelvis, biliary tract, and brain (usually glioblastoma as seen in Turcot syndrome) tumors, sebaceous gland adenomas and keratoacanthomas in Muir-Torre syndrome, and carcinoma of the small bowel.
b Presence of tumor-infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous/signet-ring differentiation, or medullary growth pattern.
LS-associated CRC has a higher incidence of several recognizable histopathologic features, such as poorly differentiated mucinous appearance, characteristic lymphocytic infiltrate, histologic heterogeneity, and signet-ring cell features. An identifiable molecular feature of LS-associated CRC is microsatellite instability (MSI). Because the MMR system is responsible for surveillance and correction of errors in DNA replication, it was discovered that sequences of DNA that are prone to accumulation of mutations, such as microsatellite repetitive sequences consisting of mononucleotide, dinucleotide, or higher-order nucleotide repeats, are not repaired if MMR genes are dysfunctional. This MSI phenotype (also known as the mutator phenotype), despite being a hallmark of LS, is also observed in 10% to 15% of sporadic CRCs. Many genes, such as TGF-β , BAX , and HDAC2 , develop mutations in microsatellite repeats in MMR-deficient CRCs. MSI is now routinely detected in tumor blocks, and a consensus conference in Bethesda established a panel of microsatellite markers (initially 5 markers, now expanded to 10) to identify MSI CRC. Based on these markers, MSI is judged to be high (MSI-H), low (MSI-L), or stable (MSS). The Bethesda criteria and later-revised Bethesda criteria (see Table 1 ) were developed to identify those patients whose CRC warrants molecular testing for MSI. However, there has been much debate as to the utility of these criteria to identify patients suitable for MMR germline testing. Recently, it has been shown that immunohistochemical (IHC) analysis of MMR protein expression can help guide further genetic testing (see the following sections).
Epigenetics
Epigenetics refers to stable changes in gene expression resulting from changes in a chromosome without alteration of the actual gene sequence. For example, on a somatic level, methylation of GC- and CpG-rich areas (usually located near the promoter regions of widely expressed genes) that are known as CpG islands contributes to gene inactivation in most cases (up to 84%) of MLH1 loss in MSI-H CRCs. It also appears that MSI CRC has more endogenous CpG methylation than MSS CRCs and this, in turn, is responsible for a greater rate of genetic change in these tumors. Tumor suppressor genes inactivated by hypermethylation include APC , BRAF , and MRE11A . Thus, detected loss of MLH1 on IHC analysis may be caused by spontaneous hypermethylation of the promoter region, resulting in MLH1 inactivation, even in a suspected LS tumor. Germline sequencing may not be required in such cases, although the challenge lies in separating these from the actual LS tumors.
Recent work on the RAF/MEK/ERK/MAPK kinase signaling pathway has shown that somatic changes in KRAS and BRAF are important in CRC tumorigenesis and therapeutics. Activating mutations in the KRAS gene are seen in 30% to 40% of CRCs, and mutually exclusive activating mutations in BRAF are present in 10% of CRCs. These mutations predict resistance to anti-epidermal growth factor receptor antibodies (eg, cetuximab). BRAF mutations are almost exclusively seen in sporadic MSI CRCs and result from somatic hypermethylation of MLH1 . Testing CRCs for somatic BRAF mutations (the most common being V600E) has therefore been suggested to rule out LS, in cases in which the tumor is MSI-H or family history suggests a genetic predisposition. KRAS mutations are more common in MSS tumors. Some recent studies have focused on genetic and epigenetic classifications, defining clinical phenotype, and determining patient outcomes in CRC. Sanchez and colleagues analyzed tumors from 391 unselected patients with CRC for MSI, CpG island methylator phenotype (CIMP), and BRAF and KRAS mutations. The investigators concluded that the population was heterogeneous. CIMP-H and MSI-H tumors were not interchangeable and both molecular subtypes had a high frequency of BRAF mutation and a low rate of KRAS mutation. Most tumors were MSS/CIMP-negative (70%). Divergent survival was seen amongst the different groups, with MSI-H/CIMP-negative tumors showing the best survival for stages I to III (stage IV was the strongest determinant of survival and was removed from the analysis). Others have confirmed these findings and have highlighted the molecular complexity of CRCs based on genetic and epigenetic profiles.
Adding further complexity to the identification of potential patients with LS, there are reports that the LS phenotype can be inherited via germline epigenetic changes affecting MLH1 or MSH2 . A heritable somatic mutation in MSH2 has been reported, resulting from germline deletions in the TACSTD1 gene, which lies directly upstream of MSH2 and encodes Ep-CAM. Such deletions result in the silencing of MSH2 by hypermethylation. It has been proposed that clinical genetics workup should include deletion analysis of the 3′ region of the TACSTD1 gene in those cases in which IHC analysis shows MSH2/MSH6 protein loss, and no mutation is found in the coding regions of the MSH2/MSH6 genes. A recent report of 331 LS-suspected patients, who had no germline MLH1 , MSH2 , or MSH6 mutation and whose tumors showed MSI-H or loss of MLH1 or MSH2 , revealed 2 patients with germline MLH1 promoter hypermethylation and 3 patients with somatic MSH2 promoter hypermethylation in their tumors caused by a germline TACSTD1 deletion. Thus germline epigenetic changes can mimic hereditary cancer syndromes. More recent data suggest that somatic methylation of MSH2 in patients with LS is common (24% of cases of LS patients with MSH2 mutations) and may serve as the second hit at the wild-type MSH2 allele.
Epigenetics
Epigenetics refers to stable changes in gene expression resulting from changes in a chromosome without alteration of the actual gene sequence. For example, on a somatic level, methylation of GC- and CpG-rich areas (usually located near the promoter regions of widely expressed genes) that are known as CpG islands contributes to gene inactivation in most cases (up to 84%) of MLH1 loss in MSI-H CRCs. It also appears that MSI CRC has more endogenous CpG methylation than MSS CRCs and this, in turn, is responsible for a greater rate of genetic change in these tumors. Tumor suppressor genes inactivated by hypermethylation include APC , BRAF , and MRE11A . Thus, detected loss of MLH1 on IHC analysis may be caused by spontaneous hypermethylation of the promoter region, resulting in MLH1 inactivation, even in a suspected LS tumor. Germline sequencing may not be required in such cases, although the challenge lies in separating these from the actual LS tumors.
Recent work on the RAF/MEK/ERK/MAPK kinase signaling pathway has shown that somatic changes in KRAS and BRAF are important in CRC tumorigenesis and therapeutics. Activating mutations in the KRAS gene are seen in 30% to 40% of CRCs, and mutually exclusive activating mutations in BRAF are present in 10% of CRCs. These mutations predict resistance to anti-epidermal growth factor receptor antibodies (eg, cetuximab). BRAF mutations are almost exclusively seen in sporadic MSI CRCs and result from somatic hypermethylation of MLH1 . Testing CRCs for somatic BRAF mutations (the most common being V600E) has therefore been suggested to rule out LS, in cases in which the tumor is MSI-H or family history suggests a genetic predisposition. KRAS mutations are more common in MSS tumors. Some recent studies have focused on genetic and epigenetic classifications, defining clinical phenotype, and determining patient outcomes in CRC. Sanchez and colleagues analyzed tumors from 391 unselected patients with CRC for MSI, CpG island methylator phenotype (CIMP), and BRAF and KRAS mutations. The investigators concluded that the population was heterogeneous. CIMP-H and MSI-H tumors were not interchangeable and both molecular subtypes had a high frequency of BRAF mutation and a low rate of KRAS mutation. Most tumors were MSS/CIMP-negative (70%). Divergent survival was seen amongst the different groups, with MSI-H/CIMP-negative tumors showing the best survival for stages I to III (stage IV was the strongest determinant of survival and was removed from the analysis). Others have confirmed these findings and have highlighted the molecular complexity of CRCs based on genetic and epigenetic profiles.
Adding further complexity to the identification of potential patients with LS, there are reports that the LS phenotype can be inherited via germline epigenetic changes affecting MLH1 or MSH2 . A heritable somatic mutation in MSH2 has been reported, resulting from germline deletions in the TACSTD1 gene, which lies directly upstream of MSH2 and encodes Ep-CAM. Such deletions result in the silencing of MSH2 by hypermethylation. It has been proposed that clinical genetics workup should include deletion analysis of the 3′ region of the TACSTD1 gene in those cases in which IHC analysis shows MSH2/MSH6 protein loss, and no mutation is found in the coding regions of the MSH2/MSH6 genes. A recent report of 331 LS-suspected patients, who had no germline MLH1 , MSH2 , or MSH6 mutation and whose tumors showed MSI-H or loss of MLH1 or MSH2 , revealed 2 patients with germline MLH1 promoter hypermethylation and 3 patients with somatic MSH2 promoter hypermethylation in their tumors caused by a germline TACSTD1 deletion. Thus germline epigenetic changes can mimic hereditary cancer syndromes. More recent data suggest that somatic methylation of MSH2 in patients with LS is common (24% of cases of LS patients with MSH2 mutations) and may serve as the second hit at the wild-type MSH2 allele.
Missense MMR mutations
There is, as yet, no consensus as to what level of evidence is sufficient to classify germline MMR mutations as deleterious. Yet unclassified mutations are referred to as variants of uncertain significance. There is significant variation with regard to age of onset, clinical phenotypes, and tumor spectrum among individuals and families with MMR gene mutations. The clinical phenotype ranges from families satisfying the highly restrictive Amsterdam criteria to familial CRC, isolated early-onset disease, and even sporadic CRC. Part of the large variation is attributable to the frequent occurrence of missense mutations that can account for up to 24% of all LS mutations (typical mutations are truncating genomic deletions, duplications, and rearrangements). Missense mutations can cause a loss of protein function and result in a phenotype similar to mutations that truncate the protein prematurely; others create proteins that retain partial function or result in non-functional proteins that are still expressed and scored as wild type on IHC analysis. There is widespread agreement that correct interpretation of the clinical significance of specific missense mutations (even with bioinformatic algorithms) is extremely challenging and complicates the genetic counseling and medical management of the families involved. A pervasive weakness of risk-prediction models for LS is that they cannot distinguish to what extent a variant is a causative deleterious mutation. Approaches to combine mutation risk prediction and missense variant classification have been developed for the BRCA1/BRCA2 genes, and it is hoped that a similar approach in CRC, such as missense variants with multivariate analysis of protein polymorphisms mismatch repair (MAPP-MMR) or align–Grantham variation-Grantham deviation (A-GVGD) combined with risk-prediction models, can help to more accurately define missense variants.
Identifying patients with LS
As the clinical management and prognosis of MMR mutation versus non mutation carriers differs significantly, it is imperative to identify those patients with LS. It is not practical to perform germline testing on all patients. The Amsterdam and broader Bethesda criteria help define a high-risk population in whom germline testing should be considered. However, these criteria have low sensitivities (40%–80%, respectively) among selected (CRC patients referred to a genetics clinic) and unselected patients.
Molecular pre-screening using MSI testing or IHC analysis of tumor tissue for MMR protein loss has been proposed as a primary screening tool for all patients with CRC. The rationale for using IHC analysis is that loss of MMR protein expression identifies the relevant deficient MMR gene in a quick, inexpensive, and reproducible manner. When a mismatch is detected, MSH2 associates with MSH6 (predominantly) and MLH1 couples with PMS2 . It follows that absence of MLH1 and PMS2 on IHC most likely represents MLH1 expression loss ( PMS2 loss is secondary). In that case, the germline workup should therefore initially focus on MLH1 . Given the accessibility of immunohistochemistry, many institutions now routinely test colon cancers (especially those patients younger than 60 years) for MMR proteins. An algorithm for the proposed institution-wide clinical evaluation for LS is summarized in Fig. 2 . If no tumor tissue is available and the suspicion for LS is strong, it is reasonable to proceed directly to MMR germline sequencing.

IHC analysis and MSI testing have limitations as screening tools. As discussed earlier, MSI tumors are seen in 10% to 15% of sporadic CRCs and most MLH1 protein loss on IHC analysis is caused by hypermethylation of the promoter in the absence of a germline mutation. A recent report from the Evaluation of Genomic Applications in Practice and Prevention Working Groups has reviewed the clinical validity of IHC analysis and MSI testing before MMR germline sequencing. For MSI, the clinical sensitivity (ability to identify LS) was 85% for MLH1 mutations, 85% for MSH2 mutations, and 69% for MSH6 mutations. The overall clinical specificity was 90% (95% confidence interval [CI], 87%–93%). The overall clinical sensitivity of IHC testing to identify LS was 83% for MLH1 , MSH2 , and MSH6 (95% CI, 65%–93%). Clinical specificity was 89% (95% CI, 68%–95%). Recent data from the United States have shown that the use of IHC analysis and MSI testing as preliminary tests for all CRCs is cost-effective (<$75,000 per life year saved compared with age-targeted testing), and universal testing using MSI or IHC detected nearly twice as many cases of LS as targeting younger patients.
In a cohort of 1566 unselected patients with CRC form the Columbus metropolitan area, IHC analysis for the 4 MMR proteins and MSI were both performed. If either preliminary test was abnormal, complete sequencing for germline mutations was performed. The overall prevalence of LS was at least 2.8% (44 out of 1566 patients). Of the 44 patients diagnosed with LS, 249 relatives of 33 probands were tested for germline mutations in MMR genes and 109 relatives tested positive, amounting to more than 3 relatives per proband with mutations. The strongest predictors of germline mutations were absence of MSH2 with or without absence of MSH6 on IHC analysis (66.7%), absence of MSH6 or PMS2 alone on IHC analysis (23.5% and 55.6%), and absence of MLH1 without MLH1 promoter hypermethylation (33.3%). Patients whose tumors had abnormal IHC results were just as likely to have LS as those patients whose tumors were MSI-H (21.4% vs 20.8%, P = .9847, χ 2 test). Younger (age <50 years) patients were more likely to have LS than patients who were older than 50 years. In this study, if tumor analysis had been limited to those patients who fulfilled the Bethesda criteria, 1 in 4 cases of LS would not have been identified.
The optimal diagnostic strategy for LS is still under debate. Researchers have recently hypothesized that BRAF testing on tumor tissue with absent MLH1 staining could identify sporadic CRC cases that would not benefit from MLH1 sequencing. Several studies have reported sensitivity and specificity of BRAF testing for sporadic CRC in patients with absent MLH1 expression on IHC analysis. Overall sensitivity was 69% (95% CI, 57%–79%) and specificity was 100% (95% CI, 93%–100%). Thus evidence suggests that BRAF V600E mutation testing and MLH1 promoter hypermethylation testing in those CRC cases with absent MLH1 protein on IHC analysis may avoid unnecessary MLH1 sequencing without a loss in LS detection.
Molecular prescreening using MSI or IHC testing assumes that tumor tissue is readily available. Also, these patients already have CRC, yet the major aim of any screening strategy is to identify LS prior to the development of cancer. In recent years, several models have been developed to predict the likelihood of carrying a germline mutation. Tumor tissue is not required for these models (except for MMRPro ) and information from personal and/or family history is an input to predict the probability of carrying a mutation. The models, using either logistic regression or Bayesian analytical approaches, can also be applied to those individuals in whom germline testing has found no mutation. To date, all of these models have been shown to outperform the revised Bethesda guidelines in terms of MMR mutation risk. Comparison of 4 models in high-risk patients and also in patients with CRC found that all 4 models could discriminate between carriers and noncarriers. As the penetrance of mutations in MSH6 and PMS2 is lower than that of MLH1 and MSH2 , it was not surprising that MSH6 mutation carriers had lower risk scores from all 4 models ( P <.015) (no model took into account mutations in PMS2 ). The investigators found that all 4 models had diagnostic utility in identifying patients with CRC from the general population who should receive further evaluation. When corrections were made for family size, the MMR predict model performed the best, achieving a sensitivity of 94% (95% CI, 73%–99%) and a specificity of 91% (95% CI, 88%–93%). This model could better identify a subset (11%) of all patients who should have additional molecular or IHC testing compared with the revised Bethesda criteria (sensitivity = 94% and specificity = 51%), which suggested additional testing for 50% of patients. As per the investigators, another advantage of all 4 models is the identification of at-risk family members who should be offered screening for colonic and extracolonic tumors while awaiting the probands’ molecular test results. All risk-prediction models are freely available on the Internet and can be easily used in the clinic to guide germline mutation testing.
Management and surveillance of patients with LS
Data on future cancer risk (penetrance) in MMR germline mutation carriers are varied and inconsistent amongst published series. One reason for this is the heterogeneity of populations studied; earlier studies focused on families with many affected members, whereas more recent studies tend to be population based. Results in both groups may be confounded by other genetic and environmental influences. Because of these inconsistencies, it is impossible to give a single summary statistic for CRC risk in MMR mutation carriers. The risk of CRC with MLH1/MSH2 mutations by age 70 years is 80% (men) and the overall risk is 10% lower for MSH6 and PMS2 mutations. Earlier studies suggested similar risks for women, but in recent studies, women have a lower risk (20%–40% lower) of developing CRC. In studies that are population based (and so avoid high-risk family bias), estimates for penetrance by age 70 years are 45% for men and 35% for women. Of note, the risk for other LS-associated cancers varies greatly depending on the gene studied; for example, endometrial cancer risk may be preferentially increased in MSH6 mutation carriers.
Based on the risk of CRC in MMR mutation carriers, surveillance guidelines have been proposed. Individuals with a known or suspected MMR mutation, or who are at risk based on a documented family mutation, should undergo colonoscopy at least every 1 to 2 years, starting at age 20 to 25 years (some have suggested starting at age 30 years in families with MSH6 mutations). Such frequent screening is advised based on the rapid transformation of polyp to carcinoma observed in the syndrome. Recent data from the Netherlands further support the efficacy of surveillance. In a cohort study of 2788 members from 146 families with LS, the standardized mortality ratio for CRC was decreased by 70% when compared with the subjects who did (n = 897) or did not (n = 1073) have surveillance colonoscopies ( P <.001). In those families without detectable MMR mutations, enhanced screening is also recommended based on data on the efficacy of colonoscopy in average- and high-risk populations. Joint guidelines from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, the American College of Radiology, and the American Gastroenterology Association (AGA) recommend that individuals with a family history of CRC or adenomatous polyps in a first-degree relative before age 60 years or in 2 or more first-degree relatives of any age (no genetic cause found) should have a colonoscopy at age 40 years or 10 years before the youngest case in the immediate family, whichever comes first. If normal, colonoscopy should be repeated every 5 years. Similar recommendations have been put forward by the AGA for individuals with a first-degree relative with CRC or advanced adenomatous polyps.
Surgery
Prophylactic colectomy is typically not performed in patients with MMR germline mutations. However, in patients with LS diagnosed with CRC, there is an increased risk of metachronous CRC (16% after 10-year follow-up ), and surgeons are faced with the decision to perform a segmental colectomy (SEG) or total abdominal colectomy (TAC) with ileorectal anastomosis (IRA). The latter has been proposed as the standard operation to decrease the risk of metachronous tumors, although this is largely based on expert opinion rather than empirical data. Quality-of-life issues can be significant after TAC. Maeda and colleagues used mathematical modeling to compare SEG and TAC in patients with LS. Predicted mean survival for young (30 years) patients was slightly better with TAC than with SEG (35.5 years vs 34.8 years.) However, when quality-adjusted life-years (QALYs) were considered, both the strategies were essentially equivalent. With advancing years, SEG became a more favorable strategy. Previous studies reported a better survival for TAC but did not account for QALYs. Thus the decision to perform SEG or TAC should be individualized based on patient factors, patient preferences, and ability of the patient to follow surveillance guidelines if SEG is chosen.
Surgical management and surveillance of other LS-associated cancers are dealt with elsewhere in this issue.
Chemotherapy
There is level I evidence supporting the efficacy of systemic chemotherapy in the treatment of stage II (high-risk only, ie, tumors causing bowel perforation or obstruction, large tumors with diffuse lymphovascular space and perineural invasion), III, and IV CRC. It is accepted that MSI-H tumors have a more favorable prognosis and are less likely to present with lymph node positive or metastatic disease. Recent data from large adjuvant clinical trials reported that MSI had more prognostic value in stage II than in stage III tumors. Until recently, MSI or MMR protein loss by IHC analysis has not routinely been used to inform decision making regarding chemotherapy treatment. Conflicting data, in retrospective studies, exist on the effect of 5-fluorouracil/leucovorin in MSI-H tumors. Irinotecan is a commonly used drug in metastatic CRC. Irinotecan binds to topoisomerase and generates double-strand DNA breaks. Homologous recombination helps to repair this DNA damage, and important in this process is the MRE11A/hRAD50/NBS1 ( MRN ) gene complex. Up to 85% of MSI CRCs tend to accumulate mutations in mononucleotide-repeat coding sequences in MRE11A and RAD50 . Cell line work has shown that such mutations render cells that are highly sensitive to irinotecan because double-stranded breaks cannot be repaired by homologous recombination. MSI subset analyses of irinotecan-based chemotherapy given in the adjuvant and metastatic setting have reported conflicting results. Some trials report an increased benefit in MSI-H tumors treated with irinotecan, and others report no difference between MSI-H and MSS cancers. The question of increased sensitivity of MSI CRC to irinotecan-based chemotherapy is not yet fully answered. Of note, there does not appear to be any difference in response or outcome to oxaliplatin-based treatment in MSI CRC. Poly(ADP)ribose polymerase (PARP) enzymes are critical components of the base excision repair (BER) pathway effecting single-strand break repair. Background endogenous single-strand breaks, if not repaired by the BER (nucleotide excision or MMR) pathways, can progress to double-strand breaks. MSI cell lines deficient in homologous repair pathways have been shown to be highly sensitive to PARP inhibitors compared with MSS cell lines. Studies are now underway in CRCs, stratified by microsatellite status, to study the efficacy of PARP inhibitors alone and in combination with chemotherapy that induces double-strand breaks (NCT00912743; www.clinicaltrials.gov ). Mammalian target of rapamycin inhibitors are also likely to be evaluated. As most MSI-H tumors do not occur in individuals with LS, these studies may or may not ultimately prove to be relevant to LS tumors.
Chemoprevention
The role of nonsteroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 ( COX-2 ) inhibitors has been well studied in patients with familial adenomatous polyposis (FAP; discussed in the following sections). In patients with LS, chemoprevention is less well studied. However, a recent randomized study of 1071 patients with LS reported no significant difference in the detection of one or more colorectal adenomas or CRC in those who took aspirin, resistant starch (undigested portion of starch that can suppress the proliferation of colonic cells), or both every day for up to 4 years. Phase I/II multicenter trials of celecoxib in patients with LS have been completed and are yet to be reported ( www.clinicaltrials.gov ; NCT00001693). Recent concern regarding the cardiovascular safety of COX-2 inhibitors has cast doubt on the future role of NSAIDs in adenoma and CRC prevention.
FAP
The second most common, but most easily recognizable, form of hereditary CRC is FAP (see Fig. 1 ). The prevalence of FAP is around 1 in 8000 individuals. This syndrome is characterized by hundreds or thousands of adenomatous polyps in the colon and rectum, beginning in the late teens or early 20s (median age of adenomas is 16 years) and progressing to CRC by 40 to 50 years of age in virtually every case. A minimum of 100 polyps are generally necessary to diagnose the classic form of FAP, whereas LS mutation carriers rarely have more than 12 polyps. Benign hamartomas are seen in the gastric fundus and body in about half of the patients and duodenal polyps in about 90% (4%–12% resulting in duodenal/ampullary carcinoma). There is also an increased risk for thyroid cancer (especially papillary), mostly in women, and young children are at risk for hepatoblastoma. CNS tumors (medulloblastomas) can be seen in the Turcot variant of FAP. The Gardner syndrome variant of FAP is characterized by cutaneous soft tissue lipomas, fibromas, epidermoid/sebaceous cysts on legs, face, scalp, and arms, osteomas (skull and mandible), supernumerary teeth, congenital hypertrophy of the retinal pigment epithelium (CHRPE), and desmoid tumors and mesenteric fibromatosis.
The gene responsible for FAP is the APC gene, a large gene with 15 exons, located at chromosome 5q21. Somatic mutations and deletions that inactivate both copies of APC are present in 60% to 80% of sporadic CRCs and adenomatous polyps. This gene encodes a large protein that acts in the Wnt pathway via several domains to regulate the cell cycle and apoptosis, stabilize the cytoskeleton, and mediate intercellular adhesion. The protein product of the APC gene has been shown to associate with proteins (catenins) that bind to the cell surface molecules (cadherins) essential for cellular adhesion. Activation of the Wnt signaling pathway via mutations in the APC gene is regarded as the initiating event in sporadic CRC. Germline mutations in genes that produce scaffold proteins, for example, AXIN1 and CTNNB1 , can lead to multiple polyps, which suggests that all elements of the Wnt signaling pathway can be considered putative CRC-causing genes because mutations in these genes may impact degradation of the key effector molecule of the pathway, β-catenin. Most mutations in the APC gene (98%) lead to protein truncation. Germline mutations in APC are inherited in an autosomal dominant fashion and are 100% penetrant. Approximately 30% occur de novo.
Variable genotype or phenotype correlations have been demonstrated in large kindred series. Severe polyposis is associated with APC mutations in codons 1250 to 1464, sparser polyps with mutations in codons 213 to 1249 and 1465 to 1597, CHRPE with mutations in codons 311 to 1444, and Gardner syndrome with mutations in codons 1395 to 1578. Two hotspots, codon 1061 and 1309 mutations, account for 11% and 13% of all germline mutations, respectively, with codon 1309 mutation being associated with a younger age of onset of CRC. Variable expressivity, for example, age of onset of CRC, has also been noted even within families with identical APC mutations, suggesting that modifier genes play a role.
Genetic Testing and Management
Gastroenterologists and surgeons may consider genetic testing unnecessary in patients with presumed FAP because diagnosis by sigmoidoscopy is clear and the prognosis remains unchanged (100% penetrance). However, determination of family members at risk through genetic testing can reduce unnecessary screening from a young age. Given the 100% penetrance for colon cancer, prophylactic colectomy is standard. Guidelines for the management of patient with FAP have been promulgated by the AGA and the National Comprehensive Cancer Network (NCCN) and should be consulted as they are updated regularly. In general, patients with APC mutations should undergo flexible sigmoidoscopy annually, starting at age 10 to 12 years or during the midteen years, with colonoscopy once polyps are detected and colectomy for dense polyposis (>approximately 20–30). Surgical options include colectomy and IRA, proctocolectomy with ileostomy, and proctocolectomy with ileal pouch–anal anastomosis (IPAA). The goal of colectomy and IRA is to preserve rectal function, and postsurgical flexible sigmoidoscopy surveillance of the rectum with endoscopic snare polypectomy and argon coagulation endoscopy ablation are suggested at 6-month intervals. A large analysis of the Danish, Swedish, Finnish, and Dutch polyposis registries revealed 47 rectal carcinoma diagnoses among 659 patients who had IRA surgery. The cumulative risk according to chronologic age was 30% at 60 years and higher in patients undergoing surgery after 25 years ( P = .0016). Of 167 individuals harboring known APC mutations, 7 had rectal cancer diagnoses. The 5-year median survival rate after rectal carcinoma diagnosis was 60%. In patients with FAP, regression of rectal polyps has been noted after IRA, suggesting possible environmental modulators of the phenotype. Upper gastrointestinal (GI) endoscopy should be performed in patients with FAP every 1 to 4 years with baseline examination by age 25 to 30 years, and their thyroid glands should also be palpated. At-risk infants may be considered for screening for hepatoblastoma (age 0–5 years). Presymptomatic individuals with a family history of FAP, but for whom mutation status is unknown, should have annual flexible sigmoidoscopy beginning at age 10 to 12 years until age 24 years, every 2 years until age 34 years, every 3 years until age 44 years, and every 3 to 5 years thereafter; colonoscopy should be considered at 10-year intervals starting at age 20 years.
Because the progression from initiation of adenoma to carcinoma can take several years, there is a window of opportunity to potentially intervene. NSAIDs and the more selective COX-2 inhibitors have demonstrated significant chemopreventive benefits in FAP (reduction in the size and number of rectal adenomas) as evidenced by at least 4 randomized trials. NSAIDs, however, have not been shown to prevent colorectal neoplasia in patients with FAP. The response to NSAIDs may be genetically determined. Despite these data, surgery is still the primary treatment of FAP. NSAID chemoprevention is an adjunct to surgery to address remaining rectal tissue, or may permit surgery to be delayed. Postmenopausal hormone replacement therapy has been suggested as a chemoprevention for colon cancer. Case reports of polyp regression after cytotoxic chemotherapy have been published. Further studies are required to determine whether cytotoxic chemotherapy may be an alternative for patients with FAP for whom colectomy is not an option or for duodenal or ampullary polyps that may be less amenable to surgery or treatment with NSAIDs.
Related posts:
 Upper Gastrointestinal Cancer Predisposition Syndromes
Upper Gastrointestinal Cancer Predisposition Syndromes
 Clinical Genetics of Hereditary Colorectal Cancer
Genome-wide Association Studies of Cancer Predisposition
Upper Gastrointestinal Cancer Predisposition Syndromes
Hereditary Genodermatoses with Cancer Predisposition
Genitourinary Cancer Predisposition Syndromes
Clinical Genetics of Hereditary Colorectal Cancer
Genome-wide Association Studies of Cancer Predisposition
Upper Gastrointestinal Cancer Predisposition Syndromes
Hereditary Genodermatoses with Cancer Predisposition
Genitourinary Cancer Predisposition Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


