Chronic Leukemias
Anupriya Agarwal
John C. Byrd
Michael W. Deininger
Chronic myeloid leukemia (CML) and chronic lymphocytic leukemia (CLL) are very different diseases and yet share important clinical features. Both are usually diagnosed in an indolent stage characterized by expansion of differentiating cells that can last for several, sometimes many years. In both, the acquisition of additional mutations promotes progression to advanced therapy-refractory disease and both are incurable with currently available drug therapy. In this chapter, we will discuss the key pathogenetic mechanisms of CML and CLL, with emphasis on recent data and potential therapeutic implications.
CHRONIC MYELOID LEUKEMIA
CML is caused by BCR-ABL, a constitutively active tyrosine kinase generated as the result of a reciprocal translocation between chromosomes 9 and 22. The annual incidence of CML is 1.3 to 1.5/105, with a slight male preponderance, but no significant differences across ethnicities. The only established CML risk factor is exposure to ionizing radiation, evident from studies in survivors of the nuclear explosions in Japan and patients exposed to thorotrast or radiotherapy. During the initial chronic phase (CP) cellular differentiation and function are largely maintained, therapy is effective and mortality is low. Without effective treatment the disease invariably progresses to blastic phase (BP), a rapidly fatal acute myeloid or lymphoid leukemia.
Pathogenesis
The first cases of what was probably CML were described by Bennett and Virchow in the mid 1840s.1 In 1960, Philadelphia cytogeneticists Nowell and Hungerford2 described a “minute” chromosome 22 in CML cells that became known as the Philadelphia chromosome (Ph). In 1973, Janet Rowley3 discovered that Ph is in fact the result of a reciprocal translocation between chromosomes 9 and 22 [t(9;22) (q34;q11)]. The genes juxtaposed by the translocation were subsequently identified as ABL (Abelson) on 9q344 and breakpoint cluster region (BCR) on chromosome 22q11 (Fig. 32.1). The next critical discoveries were that the constitutive tyrosine kinase activity of BCR-ABL is required for cellular transformation and that the clinical disease was reproducible in a murine model.5,6 According to the World Health Organization the presence of BCR-ABL in the context of a myeloproliferative neoplasm is diagnostic of CML, although the translocation is also found in a subset of patients with acute lymphoblastic leukemia (ALL) and rare cases of acute myeloid leukemia (AML).
Molecular Anatomy of the BCR-ABL Junction
The breakpoints within ABL occur upstream of exon 1b, downstream of exon 1a, or, more frequently, between the two. Regardless of the exact breakpoint location, splicing of the primary transcript yields an mRNA in which BCR sequences are fused to ABL exon a2. Breakpoints within BCR localize to one of three breakpoint cluster regions (bcr). More than 90% of CML patients and one-third of Ph+ ALL patients express the 210kDa isoform of BCR-ABL, in which the break occurs in the 5.8-kb major breakpoint cluster region (M-bcr), which spans exons e12-e16 (formerly exons b1-b5). Alternative splicing gives rise to either b2a2 (e13a2) or b3a2 (e14a2) transcripts.5 In remaining Ph+ ALL patients and in rare CML cases, the breakpoints are further upstream in the 54.4-kb minor breakpoint cluster region (m-bcr), generating an e1a2 transcript that is translated into p190BCR-ABL.7 A third breakpoint downstream of exon 19 in the micro breakpoint cluster region (μ-bcr) gives rise to an e19a2 BCR-ABL mRNA and p230BCR-ABL and is associated with neutrophilia.
Other BCR-ABL variants, including b2a3, b3a3, e1a3, e6a2 or e2a2, have been observed in isolated cases.8 Deletions flanking the breakpoints are detected in a subset of patients, which confer a poor prognosis to patients on interferon therapy, but probably not on imatinib.9,10,11 The reciprocal ABL-BCR transcript, although
detectable in approximately two-thirds of patients, does not seem to play any significant role in pathogenesis.12
detectable in approximately two-thirds of patients, does not seem to play any significant role in pathogenesis.12
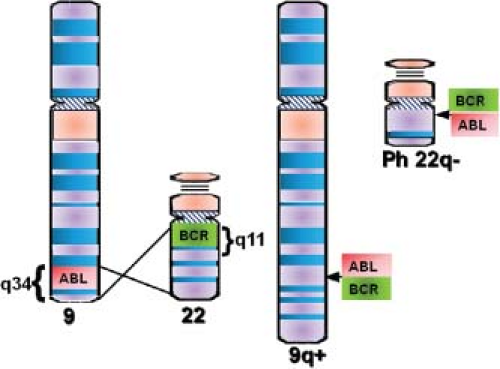 FIGURE 32.1 Schematic representation of the t(9;22) (q34;q11) translocation that creates the Philadelphia (Ph) chromosome. The ABL and BCR genes reside on the long arms of chromosome 9 and 22, respectively. As a result of the (9;22) translocation, the BCR-ABL gene is formed on the derivative of chromosome 22 (22q−, Ph chromosome), while the reciprocal ABL-BCR resides on the derivative of 9q+. |
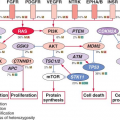
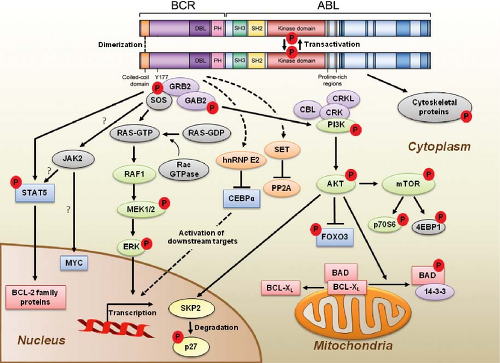 FIGURE 32.2 BCR-ABL domain structure and simplified representation of molecular signaling pathways activated in CML cells. Following dimerization of BCR-ABL, autophosphorylation generates docking sites on BCR-ABL that facilitate interaction with intermediary proteins such as GRB2. CrkL and CBL are also direct substrates of BCR-ABL that are part of a multimeric complex (purple). These BCR-ABL-dependent signaling complexes in turn lead to activation of multiple pathways whose net result is enhanced survival, inhibition of apoptosis, and perturbation of cell adhesion and migration. A subset of these pathways and their constituent transcription factors (blue), serine/threonine-specific kinases (green), cell-cycle regulatory protein (yellow), and apoptosis-related proteins (red) are shown. Several pathways were identified that are responsible for CML stem cell maintenance and/or BCR-ABL-mediated disease transformation (orange). |
Functional Domains of BCR-ABL and Kinase Activation
p210BCR-ABL contains several distinct domains (Fig. 32.2).13 The N-terminal coiled-coil domain of BCR allows BCR-ABL dimerization, which is critical for kinase activation. The p210BCR-ABL protein also retains the serine/threonine kinase and Rho guanine nucleotide exchange factor homology (Rho-GEF) domains of BCR, which are deleted in p190BCR-ABL, which may explain the differences in disease phenotype associated with the two variants. In contrast to BCR, the ABL sequence is almost completely retained, including SRC homology domains 2 and 3, the tyrosine kinase domain, a proline-rich sequence, and a
large C terminus with nuclear localization signal, DNA-binding, and actin-binding domains. The N-terminal “cap” region of ABL negatively regulates kinase activity by binding to a hydrophobic pocket at the basis of the kinase domain, which in the Ib isoform is mediated by N-terminal myristoylation. It is believed that the replacement of the cap with BCR sequences contributes to constitutive kinase activation.
large C terminus with nuclear localization signal, DNA-binding, and actin-binding domains. The N-terminal “cap” region of ABL negatively regulates kinase activity by binding to a hydrophobic pocket at the basis of the kinase domain, which in the Ib isoform is mediated by N-terminal myristoylation. It is believed that the replacement of the cap with BCR sequences contributes to constitutive kinase activation.
Signal Transduction
Numerous substrates and binding partners of BCR-ABL have been identified (Fig. 32.2). Current efforts are directed at linking these pathways to the specific phenotypic defects that characterize CML, such as increased proliferation, decreased apoptosis, defective adhesion to bone marrow stroma, and genetic instability.14 As comprehensive review of the multiple implicated pathways is beyond the scope of this chapter, we will focus on those for which strong evidence supports a ratelimiting role in disease pathogenesis.
Phosphatidylinositol-3 Kinase (PI3K)
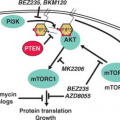
PI3K is activated by autophosphorylation of tyrosine 177, which generates a high-affinity docking site for the SH2 domain of the GRB2 adapter, which in turn recruits GAB2 into a complex that activates PI3K.15,16 Consistent with a critical role of the Y177/GRB2/GAB2 axis, mutation of tryosine 177 to phenylalanine or lack of GAB2 abrogates myeloid leukemia.16 An alternative pathway of PI3K activation is complex formation between its p85 regulatory sub-unit, CBL, and CrkL, which bind to the SH2 and proline-rich domains of BCR-ABL.17 PI3K activates the serine-threonine kinase AKT, which suppresses the activity of the forkhead O transcription factors (FOXO), thereby promoting survival.18,19 Additionally PI3K enhances cell proliferation by promoting proteasomal degradation of p27 through up-regulation of SKP2, the F-Box recognition protein of the SCFSKP2 E3 ubiquitin ligase; absence of SKP2 from the leukemia cells prolongs survival in a murine CML model.20 Another important outlet of PI3K signaling is AKT-dependent activation of mTOR, which leads to the constitutive phosphorylation of ribosomal protein p70S6 kinase and 4E-BP1, which enhance protein translation and cell proliferation.21,22
RAS/Mitogen-activated Protein Kinase (MAPK) Pathways
GRB2-mediated recruitment and activation of SOS promotes exchange of GTP for GDP on RAS.15,23 GTP-RAS activates MAP kinase, promoting proliferation. Signaling from RAS to MAPK involves the serine-threonine kinase RAF-124,25 and RAC, another GTP-GDP exchange factor.26 A crucial role for the latter is supported by the fact that lack of RAC1/2 delays BCR-ABL leukemia in a murine model.27
Janus Kinase (JAK) Signal Transducer and Activator of Transcription (STAT) Pathway
BCR-ABL activates STAT5 through direct phosphorylation or indirectly through phosphorylation by HCK, a SRC family kinase, or JAK2.28,29 Active STAT5 induces the transcription of antiapoptotic proteins like MCL-1 and BCL-XL.30 Initial experiments in mice failed to demonstrate a critical role for STAT5.31 However, it was subsequently established that the mice used in this study were not null for STAT5, but expressed an N terminally deleted protein with partially retained function. In contrast, complete lack of STAT5 abrogates both myeloid and lymphoid leukemogenesis.32
Cytoskeletal Proteins
BCR-ABL phosphorylates several proteins involved in adhesion and migration, including FAK, paxillin, p130CAS and HEF1. This and activation of RAS15,33 are thought to impair integrinmediated adhesion of CML progenitors to stroma and extracellular matrix, causing premature circulation as well as abnormal proliferation of Ph+ progenitors.34
DNA Repair
BCR-ABL impairs DNA damage surveillance by various mechanisms. For example, BCR-ABL has been shown to suppress checkpoint kinase 1 (CHK1) through inhibition of ATR35 or down-regulation of BRCA1, a substrate of ataxia telangiectasia mutated (ATM).36 Nonhomologous end-joining and homologous recombination, both critical double-strand break repair pathways, are defective in CML. BCR-ABL also up-regulates RAD51, inducing rapid but low-fidelity double-strand break repair on challenge with cytotoxic agents and induces reactive oxygen species that promote chronic oxidative DNA damage, double-strand breaks and point mutations. Lastly, telomere length decreases with disease progression from CP to BP.37
Although progress has been made to understand the extraordinary complexity of BCR-ABL signaling, a complete picture is still elusive. To overcome the limitations of investigating single pathway, quantitative proteomics is being used to establish a comprehensive picture of BCR-ABL signaling.38 These results suggest that cellular processes in CML, rather than relying on a single pathway, use integrated networks to fully realize their leukemogenic potential.
Murine Models of CML
The most commonly used murine model of CML is retroviral expression of BCR-ABL in bone marrow followed by transplantation into lethally irradiated syngeneic recipients, which develop a CML-like myeloproliferative neoplasm.39 Recently, an inducible transgenic mouse model has been developed, in which conditional BCR-ABL expression is under the control of the 3′ enhancer of the murine stem cell leukemia (SCL) gene. This model serves as a promising new tool for studying leukemogenic mechanisms in hematopoietic stem cells during disease initiation and progression.40 Lastly, xenograft models use various strains of immunodeficient mice for engraftment of primary CML cells.41 The problem with xenograft models is that the engraftment of CP cells is low, probably because of their compromised interactions with the microenvironment as a result of species differences in cytokines and adhesion molecules. More promising results have been obtained by injecting CML cells directly into the livers of newborn mice.42
CML Stem Cell
The origin of CML in a pluripotent hematopoietic stem cell (HSC) was elegantly demonstrated in the late 1970s in studies showing clonality of granulocytes, erythrocytes, and platelets of female patients heterozygous for glucose-6-phosphate dehydrogenase, a polymorphic X-chromosomal gene.43 BCR-ABL does not confer self-renewal, implying that Ph must be acquired by an HSC already endowed with this capacity.44 For unknown reasons the main cellular expansion occurs in the progenitor cell compartment, while at least initially the majority of HSC are Ph-negative.45 Serial xenograft studies have shown that CML leukemia stem cells (LSCs) reside within the quiescent CD34+ 38− fraction of bone marrow cells. Significant progress has recently been made by the identification of the IL-1 receptor-associated protein (IL-1RAP) as a surface marker specifically expressed on CD34+38− CML HSC.45a If confirmed, this will enable studies into the biology of these cells. Several genes were shown to have a critical role for leukemia-initiating cells maintenance in CML, including promyelocytic leukemia,46 the hematopoietic-specific Rac2 GTPase,47 smoothened, which is an essential component of the hedgehog pathway,48 and β-catenin. The fact that these genes are also critical for maintenance and self-renewal of normal HSCs may be an obstacle to exploiting them as therapeutic targets.
Progression to Blastic Phase
Disease progression is believed to be due to the accumulation of molecular abnormalities that lead to a loss of terminal differentiation capacity of the leukemic clone, which however continues to depend on BCR-ABL activity. BCR-ABL mRNA and protein levels are higher in BP than in CP cells, including CD34+ granulocyte macrophage progenitors (GMPs), which are expanded in BP.49 Another mechanism that enhances BCR-ABL activity in BP is inactivation of PP2A through up-regulation of SET, which in turn inhibits the PP2A phosphatase and its substrate protein tyrosine phosphatase I (SHP1). As SHP1 normally promotes BCR-ABL degradation through dephosphorylation, its reduced activity stabilizes BCR-ABL.50
The most striking feature of BP, the loss of differentiation capacity, suggests that the function of key myeloid transcription factors must be compromised. Occasionally, the differentiation block can be ascribed to mutations that result in the formation of dominant-negative transcription factors such as AML1-EVI or NUP98-HOXA9, which block differentiation or favor preferential growth of immature precursors.51,52,53 Isolated cases of myeloid transformation have been associated with the acquisition of core binding factor mutations typical of AML. A more universal mechanism appears to be the BCR-ABL-induced down-regulation of CCAAT/enhancer binding protein-α (CEBPα) through the stabilization of the translational regulator heterogeneous nuclear ribonucleoprotein E2 (hnRNP E2), which is low or undetectable in CP but readily detectable in BP CML.54 Interestingly, dominant-negative mutations of CEBPα are fairly common in AML,55 but rare in CML-BP.56
Aberrant activation of Wnt/β-catenin signaling is believed to contribute to CML progression by conferring self-renewal capacity to GMPs. Activated β-catenin regulates self-renewal by undergoing translocation to the nucleus where it interacts with lymphoid enhancer factor/T-cell factor and regulates the transcription of genes such as Myc and Cyclin D1.57 The acquisition of self-renewal by GMPs is expected to greatly increase the pool of LSCs in BP. Interestingly, expression microarray studies have implicated β-catenin activation not only in disease progression but also in resistance to tyrosine kinase inhibitors, supporting the view that drug resistance and disease progression share a common genetic basis.58,59 This has implications for prognostication as well as for the development of strategies to prevent progression and overcome resistance.
Conclusions
BCR-ABL orchestrates an integrated network of signaling pathways that upend the physiological control of proliferation, cell death, DNA repair and microenvironment interaction, and lead to the clinical phenotype of CML.
Cooperation with additional genetic events that accumulate over time inevitably leads to BP and drug resistance. Although significant progress has been made toward understanding transformation and disease progression, much remains to be learned. The availability of genome-wide scanning tools will undoubtedly accelerate this process and hopefully lead to the discovery of new therapeutic targets to eliminate CML LSC, overcome drug resistance unless effective therapy is initiated early on, and improve the prognosis of patients whose disease has progressed to BP.
Cooperation with additional genetic events that accumulate over time inevitably leads to BP and drug resistance. Although significant progress has been made toward understanding transformation and disease progression, much remains to be learned. The availability of genome-wide scanning tools will undoubtedly accelerate this process and hopefully lead to the discovery of new therapeutic targets to eliminate CML LSC, overcome drug resistance unless effective therapy is initiated early on, and improve the prognosis of patients whose disease has progressed to BP.
CHRONIC LYMPHOCYTIC LEUKEMIA
CLL is one of the most common types of leukemia in adults and has a relatively consistent immunophenotype including dim-surface immunoglobulin expression, CD19, CD20, CD23 along with the pan T-cell marker CD5.60 The impact on overall survival in young and elderly patients with CLL is quite substantial. Patients with a diagnosis under the age of 50 have a median expected lifespan of 12.3 years, which compares to 31.2 years in an age-matched control group.61 Although younger patients have poor outcome and shortened survival with CLL, several recent studies have also identified elderly patients as a high-risk group for poor survival following treatment.62,63,64,65 A small subset of CLL patients have indolent disease for many years and do not require therapy or intervention. Improving our understanding of the origin, biology, and progression of CLL will best help define both risk stratification of patients and also identify potential new treatments for this disease.
Origin of CLL
Attempts to identify the origin of CLL with respect to a normal B-cell counterpart has also occurred and remains a controversial area.66,67,68 Unlike most other B-cell lymphomas and leukemia with the exception of mantle cell lymphoma, CLL expresses typical mature B-cell markers with coexpression of CD5. This prompted many to hypothesize that CLL may be derived from CD5+ B cells whose immunoglobulin (Ig)VH is unmutated. However, the overall phenotype of CLL with expression of CD5, CD23, and CD19, and low levels of sIgM or IgD is not observed in any other type of normal B-cell counterpart. Additionally, investigators identified that approximately 40% of CLL cases are IgVH unmutated, whereas the remainder are IgVH mutated.69,70 These two groups also were shown to have distinct clinical features, prompting the hypothesis that CLL may represent two distinct diseases.69,70
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


