Chronic Autoimmune Thyroiditis
Anthony P. Weetman
Chronic autoimmune thyroiditis is the original paradigm for autoimmune diseases in general. Only foreign proteins were considered to be antigens until Witebsky and Rose demonstrated that rabbits immunized with a saline extract of homologous thyroid tissue and Freund’s adjuvant produced thyroid antibodies and had lymphocytic infiltration in their thyroid glands (1). In the same year, 1956, Doniach and Roitt reported that many patients with chronic goitrous (Hashimoto’s) thyroiditis had high serum concentrations of thyroglobulin (Tg) antibodies (2). A year later, a second autoantigen was identified in the microsomal fraction of thyroid homogenates; it proved to be thyroid peroxidase (TPO) (3). We know now that other components of thyroid tissue, including the thyrotropin (TSH) receptor and, less commonly, the sodium/iodide cotransporter and pendrin, can serve as autoantigens.
The term autoimmune thyroiditis encompasses several different clinical entities whose interrelationships remain unclear (Table 35.1). The most important are chronic goitrous thyroiditis and chronic atrophic thyroiditis. The pathology of these disorders, considered in detail in Chapter 15, consists of varying degrees of lymphocytic infiltration, fibrosis, and loss of follicular epithelium; occasionally some follicular hyperplasia is seen if the thyroid gland is enlarged. Repeat biopsies, performed at intervals of up to 20 years, reveal little alteration in thyroid histology, even in patients treated with thyroxine (T4) (4). Although there is no evidence that goitrous thyroiditis always precedes atrophic thyroiditis, the atrophic and goitrous variants share many clinical and biochemical, as well as pathologic features, suggesting that their pathogenesis overlaps. This concept of a disease continuum is supported by the unimodal distribution of thyroid volume in autoimmune hypothyroidism (5).
Predisposition to Chronic Autoimmune Thyroiditis
Autoimmune diseases usually occur when there is a failure of T-cell tolerance as a result of a combination of genetic and non-genetic factors (Table 35.2). The importance of environmental factors is illustrated by the dramatic rise in Hashimoto’s thyroiditis that has been reported in Italy, for reasons which are unclear and not related to any established precipitant (6). The high prevalence of chronic autoimmune thyroiditis in women is partly related to the effects of sex hormones, and is best demonstrated by manipulation of estrogen and androgen concentrations in experimental autoimmune thyroiditis and other animal models of autoimmune disease (7). Postpartum thyroiditis (see Chapter 56) also may result from hormonal effects on the immune system and is an important predictor of subsequent hypothyroidism. An alternative explanation for the effect of pregnancy in thyroid autoimmunity is the occurrence of intrathyroidal fetal microchimerism (8). Fetal cells may provoke an intrathyroidal alloimmune reaction that would be especially pronounced in those with pre-existing subclinical autoimmune thyroiditis.
A role for stress is not apparent in chronic autoimmune thyroiditis, possibly because the disease evolves over such a long period, but stress-related variations in cortisol secretion could affect susceptibility, as suggested by observations in animal models of thyroidtis (see later). Smoking protects against the development of thyroid antibodies and autoimmune hypothyroidism (9).
Normal gut microflora are required for the appearance of experimental autoimmune thyroiditis that develops after thymectomy and irradiation in rats (10) and serologic evidence of thyroid autoimmunity is greater in children who live in areas of high prosperity and standards of hygiene (11). These observations are in keeping with the ‘hygiene hypothesis,’ whereby decreased exposure to common infections during development skews the immune system toward the development of autoimmunity, for instance by deviating away from Th1 responses toward Th2 responses (see later) (12). There is an increase in autoimmune thyroid disease in patients with the congenital rubella syndrome (13), and hepatitis C virus infection has also been associated with autoimmune thyroiditis (14). On the other hand, chronic autoimmune thyroiditis is not a sequel of subacute granulomatous thyroiditis, which is caused by a viral infection.
Table 35.1 Types of Autoimmune Thyroiditis | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 35.2 Roles of Genetic and Non-Genetic Factors in Autoimmunity | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Observations in animals provide the most compelling evidence that a high dietary iodine intake can exacerbate thyroid autoimmunity. A high iodine intake increases the frequency of spontaneous thyroiditis in rats and Obese strain chickens, as does a normal amount given after a period of iodine restriction (15). The effects of iodine supplementation on thyroid autoimmunity in humans are contradictory, probably due to the existence of immunoregulatory mechanisms that render any effects transient, and also genetic and environmental differences. Nonetheless, iodine administration can increase the frequency of high serum thyroid antibody concentrations and lymphocytic thyroiditis in patients with goiter and can increase the frequency of hypothyroidism (16,17).
In patients with cancer or chronic hepatitis who have subclinical chronic autoimmune thyroiditis, treatment with interferon-α (IFN-α) or interleukin-2 (IL-2) worsens the autoimmune process and may cause transient thyrotoxicosis, hypothyroidism, or both, and sometimes permanent hypothyroidism (14,18). The frequency of thyroid antibodies was transiently increased in children exposed to fallout after the Chernobyl nuclear reactor accident in Ukraine (19). Lithium
exacerbates autoimmune thyroiditis, whereas selenium supplementation may reduce thyroid antibody levels (20,21).
exacerbates autoimmune thyroiditis, whereas selenium supplementation may reduce thyroid antibody levels (20,21).
Pathogenesis of Chronic Autoimmune Thyroiditis
Antigen Presentation
The initial event in the autoimmune process is presentation of antigen, in which an antigen is taken up by cells and processed into peptide epitopes, and the peptides then combine with major histocompatibility complex (MHC) class II molecules on those cells to form bimolecular complexes (22) (Fig. 35.1). The binding of these complexes to T-cell antigen receptors leads to T-cell activation and the release of cytokines, which, for instance, can provide help for B cells to make antibodies. To stimulate T cells, particularly naïve T cells not previously exposed to antigen, costimulatory signals are required, the best characterized of which is a family of proteins called B7 found on antigen-presenting cells (23). These proteins bind to CD28 receptors on T cells to augment or induce T-cell activation by the complexes of MHC class II and peptide (antigen) molecules.
By binding, instead, to cytotoxic lymphocyte antigen-4 (CTLA-4) expressed on activated T cells, B7 can terminate T-cell activation. Both CD28 and CTLA-4 play critical roles in the function of Treg cells. Other costimulatory signal receptors exist such as CD40 (24) and targeting such costimulators may offer the potential to treat autoimmune disease.
By binding, instead, to cytotoxic lymphocyte antigen-4 (CTLA-4) expressed on activated T cells, B7 can terminate T-cell activation. Both CD28 and CTLA-4 play critical roles in the function of Treg cells. Other costimulatory signal receptors exist such as CD40 (24) and targeting such costimulators may offer the potential to treat autoimmune disease.
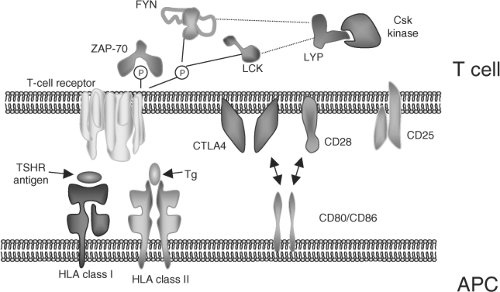 Figure 35.1 Major molecular interactions in antigen presentation. In a typical antigen-presenting cell (APC), such as a dendritic cell, protein antigen is processed into a peptide epitope that binds to the polymorphic region of MHC (termed HLA in man) class I and II molecules. The interaction with TSHR and Tg is shown: Either antigen can be presented by class I or class II. This complex of MHC and epitope is recognized by the heterodimeric T-cell antigen receptor, composed of an α- and a β-chain. Full activation of the T cell also requires a costimulatory signal, such as that supplied by CD80/CD86 (or B7-1/B7-2) through CD28. If these costimulatory molecules engage instead with cytotoxic lymphocyte antigen-4 (CTLA-4) on the T cell, the result is anergy, this pathway being responsible for the termination of an immune response. T-cell signaling is further regulated by other molecules including CD25 (IL-2 receptor) and the involvement of lymphoid phosphatase (LYP) encoded by the PTPN22 gene in downstream signaling. (Reprinted from: Zeitlin A, et al. Genetic developments in autoimmune thyroid disease: An evolutionary process. Clin Endocrinol (Oxf) 2008;68:671, with permission.) |
Most antigen-presenting cells such as dendritic cells constitutively express MHC class II molecules and costimulatory molecules. Normal thyroid cells do not express class II molecules, but those from patients with chronic autoimmune thyroiditis do (25). A close spatial correlation exists between class II molecules on thyroid cells and IFN-γ–containing T cells in thyroid tissue (26); this T-cell–derived cytokine is the only known initiator of class II expression by normal thyroid cells in vitro. In animals with experimental autoimmune thyroiditis, MHC class II–positive thyroid cells only appear late in the disease, after T-cell infiltration (27). Therefore, MHC class II expression on thyroid cells is probably the consequence rather than the cause of thyroid autoimmunity, and as a result, antigen presentation by thyroid cells is likely to be important only in the perpetuation of the autoimmune response.
Table 35.3 T-Cell Mechanisms Involved in Immunologic Self-Tolerance | |||||||
|---|---|---|---|---|---|---|---|
|
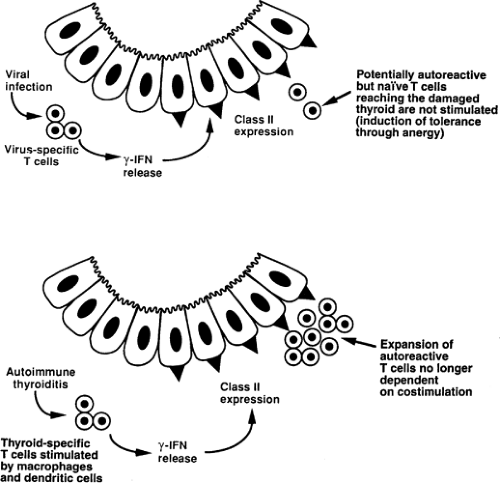 Figure 35.2 Alternative outcomes after expression of MHC class II molecules by thyroid cells. Naïve T cells undergo peripheral tolerance through anergy because the thyroid follicular cells do not supply costimulatory signals. However, T cells that have already encountered autoantigen presented by a macrophage or dendritic cell may be stimulated in the absence of further costimulation, exacerbating the autoimmune response. IFN, interferon. |
Class II–positive thyroid cells do not stimulate T cells that require B7, although T cells not requiring costimulation via B7 can be activated (28). Thus, in the absence of B7 expression, thyroid cells will not stimulate naïve, autoreactive T cells. Instead, these T cells might actually acquire tolerance through the operation of anergy (29), which can be viewed as a protective mechanism (Table 35.3). Once T cells have encountered an appropriately presented antigen (for instance, presented by a dendritic cell), they are less dependent on costimulation for subsequent activation and therefore could respond to class II–positive thyroid cells. This explains the fact that transgenic expression of class II MHC molecules on thyroid cells in mice does not cause spontaneous thyroiditis, but does increase its severity once thyroiditis is induced by immunization (30).
Thus, the extent of antigen presentation by thyroid cells in autoimmune thyroiditis should depend at least in part on the timing of the appearance of class II molecules on these cells. Prompt appearance of class II molecules on thyroid cells, e.g., in response to a viral infection, could be an important protective mechanism because this might render potentially autoreactive T cells anergic (Fig. 35.2). However, in chronic autoimmune thyroiditis, these T cells are presumably first activated by dendritic cells and may then be further activated by class II–expressing thyroid cells, depending on their requirement for costimulation.
T-cell Responses
Phenotypic changes in peripheral blood and thyroid-infiltrating T cells provide only indirect information on T-cell function in chronic autoimmune thyroiditis. In the circulation, the number of activated T-cell–expressing HLA-DR is increased (31) and the number of CD8 cells is decreased, but only during active thyroid damage (32). Of more direct relevance is the phenotype of T cells infiltrating the thyroid; these are mainly CD4 T cells that express activation markers such as MHC class II molecules (33).
In some animal models of autoimmune disease the T-cell response is clonally restricted in the early phase of the disease, as marked by the utilization of particular variable (Vα) region genes encoding the T-cell antigen receptor of pathogenic T cells, although with time the immune response spreads, so that the autoantigens are recognized by multiple T-cell antigen receptors (34). There is no evidence for restriction of Vα gene usage by recently activated, IL-2 receptor-positive intrathyroidal T cells from patients with Graves’ disease or chronic autoimmune thyroiditis, although a limited degree of restriction in the CD8 T-cell population was found in patients with goitrous thyroiditis (35,36). The role of these cells is unclear, but such restriction is compatible with the expansion of T cells that may be involved in thyroid-cell destruction. Moreover, in experimental autoimmune thyroiditis, marked restriction in V gene usage is not apparent even in early thyroiditis (37).
Circulating and intrathyroidal T cells from patients with chronic autoimmune thyroiditis are weakly stimulated in vitro by Tg and TPO (38). Attempts have been made using synthetic peptides or recombinant fusion proteins to identify the T-cell epitopes within TPO; on the basis of these studies, the epitopes of TPO seem to cluster between residues 415 and 589 of the protein (39). Although immunodominant domains have been defined for Tg and TPO (40,41), the overall T-cell response to these autoantigens is heterogeneous. These results are compatible with a polyclonal T-cell response that has spread to involve multiple epitopes, some of which are “cryptic” in the sense that they are recognized by the immune system only during the course of an autoimmune response. Furthermore, TPO seems to be processed differently by thyroid follicular cells and professional antigen-presenting cells, and T-cell clones respond differently to peptide fragments of TPO presented by the two types of cells (42); these variations could play an important role in the spreading of the autoimmune response.
A defect in immune suppression [now called immune regulation and mediated by T regulatory (Treg) cells] has been proposed for decades to play a major role in the pathogenesis of autoimmune thyroiditis (43). Some of the best evidence supporting the existence of these cells comes from animal models of experimental autoimmune thyroiditis, discussed further
below. Several in vitro techniques have been used to try to identify a defect in immune regulation in patients with autoimmune thyroiditis, including the migration inhibition assay and inhibition of mitogen-dependent autoantibody production (44,45). The results suggested that a subset of circulating T cells from normal subjects inhibits the function of T cells from patients with chronic autoimmune thyroiditis, and that cells of this subset are lacking in the patients. However, the physiologic relevance of these assay systems has been questioned, because the systems involve coculture of allogeneic cells and because such effects are not always antigen specific (46). The effects also do not exactly correspond with those we now expect of Treg cells, but there is no doubt from animal studies of the importance of these cells in self-tolerance to thyroid antigens (47). The phenotypic characterization of human Treg cells has allowed identification of large numbers of such cells in the thyroid infiltrate in Hashimoto’s thyroiditis, although they appear to be unable to downregulate the immune response effectively (48).
below. Several in vitro techniques have been used to try to identify a defect in immune regulation in patients with autoimmune thyroiditis, including the migration inhibition assay and inhibition of mitogen-dependent autoantibody production (44,45). The results suggested that a subset of circulating T cells from normal subjects inhibits the function of T cells from patients with chronic autoimmune thyroiditis, and that cells of this subset are lacking in the patients. However, the physiologic relevance of these assay systems has been questioned, because the systems involve coculture of allogeneic cells and because such effects are not always antigen specific (46). The effects also do not exactly correspond with those we now expect of Treg cells, but there is no doubt from animal studies of the importance of these cells in self-tolerance to thyroid antigens (47). The phenotypic characterization of human Treg cells has allowed identification of large numbers of such cells in the thyroid infiltrate in Hashimoto’s thyroiditis, although they appear to be unable to downregulate the immune response effectively (48).
Treg cells play a major role in the ability of the mother to tolerate paternal antigens expressed by the fetus during pregnancy. It is likely that the amelioration of thyroid autoimmunity during pregnancy is due to this increase in Treg function, because Treg cells are capable of linked suppression, i.e., they are able to downregulate immune responses to a variety of antigens besides that inducing the response (49). Postpartum exacerbation of thyroiditis may be a consequence of reconstitution of the normal immune milieu and disordered Treg function, as well as the hormonal and microchimerism effects mentioned previously (49).
The particular cytokines released by subsets of antigen-stimulated T-helper (Th) cells confer another type of regulatory function, with the cytokine products of the Th1 subset (cells involved primarily in cell-mediated immunity and autoimmune diseases like multiple sclerosis) suppressing Th2 cells (cells involved primarily in atopy, antibody-mediated immunity, and autoimmune diseases like Graves’ disease) and vice versa (50). Stimulation of Th2 responses in animal models of Th1-mediated autoimmunity results in deviation of the immune response away from the more harmful Th1 responses (51). On the other hand, there is evidence that an atopic (Th2-associated) phenotype may increase the risk of Th2-mediated autoimmune disease developing and this explains the association of various markers of atopic disease with relapse of Graves’ disease (52). Moreover, a reduction in Th1 responses during infancy as a consequence of improved hygiene may be one of the factors underlying the recent increase in Th2-mediated allergic and autoimmune disorders (12). It is also now clear that dendritic cells and the innate immune system have a fundamental role in controlling both the type of Th cell induced by an antigen and the induction of Treg cells, but so far defects in this limb of the immune response have not been identified as a cause of autoimmune thyroiditis (53).
A third T-cell subset, secreting the cytokine IL-17 and hence termed Th17 cells, has been implicated in attracting leukocytes to sites of inflammation, through the ability of IL-17 to induce chemokine production, and Th17 cells may also be important in mediating tissue injury (54). There is an increased differentiation of Th17 cells and enhanced Th17 cytokine production in autoimmune hypothyroidism, suggesting a role for these cells in disease pathogenesis (55). Th17 cells have also been shown to be critical effector cells in the pathogenesis of iodine-induced experimental autoimmune thyroiditis mice (56).
B-cell Responses
The laboratory assessment of thyroid antibodies in patients with chronic autoimmune thyroiditis is considered in detail in Chapter 13A. The range of heavy- and light-chain combinations of TPO and, to a lesser extent, Tg antibodies, is limited, which could be a result of the genetic control of thyroid antibody production (57,58). Immunodominant regions on the TPO and the Tg molecule have been defined, although it remains unclear why such dominance is a feature of the interaction with autoantibodies in contrast to antibodies induced by exogenous antigens (59). Cloning of TSH receptor antibodies has demonstrated that stimulating antibodies bind to the extracellular domain of the receptor in a very similar manner to TSH (60). Of particular relevance to autoimmune hypothyroidism is the function of TSH receptor–blocking antibodies. The only cloned blocking autoantibody so far binds to the same core regions of the TSH receptor molecule as TSH and stimulating antibodies, albeit with less interaction with the C-terminus of the receptor (61).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


