Autosomal dominant syndromes comprise the majority of families with single-gene disorders that convey an increased risk of cancer in both childhood and adulthood. The features of autosomal dominant inheritance include equal transmission of a heterozygous mutation from the father or mother to a son or daughter. Often, there is a multigenerational pattern, and a variable expression of the disorder within a family, with “skipped” generations (at the phenotypic level) because of incomplete penetrance.
Penetrance is defined as the probability that a person inheriting the mutation will develop the disease in question. Although autosomal dominant inheritance is described on the basis of this segregation of cancer within families, for childhood cancers susceptibility syndromes, the affected child is often the first person in the family with the disease. This situation results from the child carrying a
de novo heterozygous mutation in the dominant cancer susceptibility gene. For example, in the next section we review that 80% of children with bilateral retinoblastoma do not have a family history of the disease. As genomic technologies including whole-exome and whole-genome sequencing have evolved, there has been increasing recognition of children with hereditary forms of cancer even in the absence of family history.
Retinoblastoma
Much of our knowledge of autosomal dominant cancer families was gained from the study of retinoblastoma. In a landmark paper, Knudson hypothesized that bilateral retinoblastoma represented the hereditary form, and those patients had already acquired one “hit” or mutation.
40 The best statistical model consistent with his data indicated that the bilateral form required only one additional hit after birth but that the unilateral form required two hits.
The most striking clinical features of autosomal dominant cancer predisposition disorders are those initially observed by Knudson: hereditary forms of retinoblastoma present earlier and with a greatly increased percentage of bilateral and multiple primary tumors (reviewed in Lonser
23). Importantly, these patients are not exclusively bilateral, and variable expressivity results in approximately 15% of patients with unilateral retinoblastoma carrying a constitutional mutation. An even milder form, retinoma or retinocytoma, which spontaneously regress, can also be seen in apparently unaffected adults.
41 Approximately 10% of people with a germline mutation in
RB1 do not develop retinoblastoma, that is, incomplete penetrance.
23 However, the penetrance varies among families, with specific mutations (often missense changes or splice abnormalities) resulting in mutation carriers having a higher likelihood of not developing retinoblastoma or having unilateral (as opposed to bilateral) disease. These types of families are said to demonstrate attenuated or low-penetrant retinoblastoma.
Individuals carrying germline mutations in the
RB1 gene are at increased risk for development of other primary tumors, including osteosarcoma and malignant melanoma (both within and outside the radiation field) in childhood and leiomyosarcomas in adults. In a UK cohort of long-term survivors of bilateral retinoblastoma, there was a 48% risk of developing a second neoplasm by age 50.
42 Further follow-up of this cohort (up to age 84) identified a 68% cumulative incidence of second cancers including many epithelial cancers, for example, lung cancer, at later ages as well as leiomyosarcomas which occur almost entirely in the retroperitoneum, bladder, and uterus with equal distribution of the sexes.
43 Few individuals in the UK cohort received radiation therapy confirming that there is a significant risk of second primary cancers in all bilateral retinoblastoma patients. Data from a US cohort looking at cumulative cancer mortality (as opposed to incidence) identified 25% and 1% risk for hereditary and nonhereditary retinoblastoma, respectively.
44 Analysis of this long-term US cohort also demonstrates that the addition of alkylating agents in children <1 year of age increased the risk of leiomyosarcoma.
45 It is difficult to predict the outcomes from contemporary treatments. At 11 years of follow-up, 4% of patients with hereditary retinoblastoma receiving chemotherapy regimens consisting of carboplatin, vincristine, and etoposide demonstrated second malignancies.
46 A study comparing retinoblastoma patients who received proton therapy with those receiving photon therapy (follow-up of 5 to 10 years) demonstrates a decreased risk of second cancers.
47The gene mutated in retinoblastoma,
RB1, was identified on the basis of rare patients with cytogenetically visible deletions at 13q14.
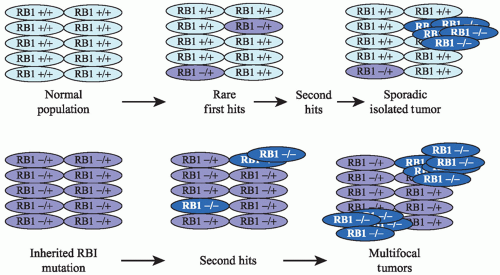
48 Molecular studies confirmed Knudson’s two-hit hypothesis. Retinoblastoma requires loss of both copies (i.e., two hits) of the
RB1 gene for a tumor to develop (
Fig. 2.1). In the familial form, a mutation in one
RB1 gene is inherited, and therefore all the cells in the body have only one normal allele. If during development that normal copy is mutated or lost, then cell cycle control is disrupted and retinoblastoma can develop. The most common mechanisms by which the second copy is lost are loss of the whole chromosome, large deletions, and gene conversion normally resulting in loss of heterozygosity (LOH) for markers near the
RB1 locus. Silencing of the gene by epigenetic methylation of the
RB1 promoter can also occur. In the sporadic form of retinoblastoma,
mutation, silencing, or loss of both
RB1 genes must occur in the same somatic retinal cell for retinoblastoma to develop.
Although bilateral retinoblastoma results from constitutional mutations in the
RB1 gene, 80% will have no family history of retinoblastoma and result from
de novo mutation in the
RB1 gene. Surprisingly, parents of a child with bilateral retinoblastoma who have normal eye exams retain a 6% risk to have a second affected child due to the
de novo mutation occurring in either the mother’s or father’s germline with a variable percentage of the eggs or sperm carrying the mutation (germline mosaicism).
49 Therefore, all siblings of children with bilateral retinoblastoma should be evaluated by genetics and have ophthalmologic surveillance beginning at birth until the genetic status is clarified.
Several different molecular approaches are used to analyze the tumor and blood specimen of retinoblastoma patients to identify point mutations scattered throughout the gene, intragenic or whole-gene deletion, and promoter methylations.
23 With extensive testing, clinical laboratories can identify the causative mutation in about 95% of bilateral cases where a blood sample is tested directly. Recent studies suggest that the remaining 5% of patients are most likely mosaic for the causative mutation with too few blood cells containing the mutation to be detected.
49 Patients with a negative family history and unilateral disease have only a 15%
a priori chance of having a germline
RB1 mutation. A negative comprehensive
RB1 mutation test from a blood sample from a unilateral patient will reduce this residual risk of hereditary disease to less than 1%. This risk can be further clarified by directly comparing RB1 results from tumor (when available) and blood.
23 Examples of genetic test results and interpretation for unilateral retinoblastoma patients are shown in
Table 2.3.
Genetic evaluation and testing is recommended for all retinoblastoma patients to first determine the hereditary nature and
RB1 mutation status of the proband’s disease (whether unilateral or bilateral) and the need for retinoblastoma surveillance of at-risk relatives. Family members are offered familial
RB1 mutation testing, a simplified test that is targeted to the specific
RB1 mutation identified in the family, for any at-risk relative. For example, in work from Texas Children’s Hospital, 48 at-risk relatives of patients with documented hereditary retinoblastoma underwent genetic testing that revealed six positive individuals who require retinoblastoma surveillance and 42 relatives who were negative and thus did not require retinoblastoma surveillance and are not at risk of having children at-risk, substantially decreasing costs.
50,51 If prenatal testing is not pursued, then any at-risk infant should have a careful eye examination within the first few weeks of life and a blood sample sent for analysis of the specific
RB1 mutation found in the affected member of the family. Only those infants that carry the mutation need subsequent surveillance for retinoblastoma (see chapter on retinoblastoma). For adult long-term survivors of unilateral retinoblastoma, a positive test of a blood sample is found in approximately 12% of cases and is informative of a hereditary form of Rb and substantial risk to future children. Conversely, if comprehensive
RB1 analysis of the blood is negative, then there is approximately a 0.5% to 1% residual risk for each offspring to develop retinoblastoma. Genetic testing of unilateral pediatric patients can be particularly informative for parents. If it can be documented that the child does not carry a constitutional
RB1 mutation then (1) the child is not at substantial risk for secondary malignancies, (2) radiation therapy is associated with less hazard, and (3) the parents have a negligible risk of having another child with retinoblastoma. The child with unilateral retinoblastoma may carry some residual risk of having an affected child given the possibility of mosaicism.
49 A coordinated team of oncologists, ophthalmologists, pathologists, and genetics professionals facilitates the delivery of optimal care for families with a child diagnosed with retinoblastoma.
Inherited TP53 Mutations, the Li-Fraumeni Syndrome, and Its Variant Phenotypes
In 1969, an inherited cancer predisposition syndrome was reported by Li and Fraumeni on the basis of characterization of four families in which at least two cases of sarcoma occurred in early life.
52 The list of LFS tumors includes premenopausal breast cancer, brain tumors, leukemias, adrenocortical carcinomas (ACC), gastric cancer, lymphoma, CPC, colorectal cancer, and possibly early onset lung cancer.
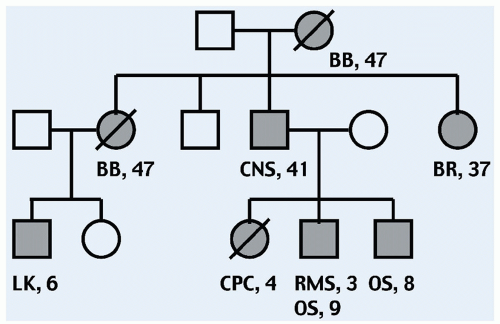
53 “Classic” LFS is defined by a proband with sarcoma diagnosed below age 45 years, who has a first-degree relative with any cancer below 45 years, plus another first or second-degree relative with either any cancer below 45 years or a sarcoma at any age. An example of an LFS pedigree is shown in
Figure 2.2. The criteria for families that do not quite meet classic criteria, termed LFS-Like (LFS-L), are generally accepted to include those outlined by the revised Chompret criteria
54 to include all children with ACC or CPC regardless of family history; a family in which the proband has multiple tumors, two of which are classical LFS tumors and the first occurred before age 46 years; or a family in which the proband has a characteristic LFS tumor diagnosed below age 46 and has at least two first- or second-degree relatives with an LFS component tumor (other than breast cancer if the proband had breast cancer).
TP53 mutation carriers exhibit greater than 80-fold increased risk of developing multiple synchronous or metachronous non-therapy-induced neoplasms.
55 In particular, the overall relative risk of occurrence of a second cancer was 5.3
(95% CI = 2.8-7.8), with a cumulative probability of second cancer occurrence of 57%.
In 1990, Malkin and colleagues detected heterozygous point mutations in the
TP53 gene in constitutional DNA in all of five LFS kindreds.
56 However, numerous subsequent studies have shown that only 60% to 80% of “classic” LFS families harbor germline
TP53 mutations (see, for example, in
Ref. 57) while the majority of LFS-L families do not have detectable
TP53 mutations (see, for example, in
Ref. 54). Mutations occur throughout the
TP53 gene, though they are primarily confined to highly conserved regions. Intragenic deletions of the
TP53 gene have been reported in a subset of families that had negative sequencing studies.
58 In some patients, the tumors do not demonstrate LOH and the
TP53 mutation is thought to act in a dominant negative form.
59The cancer phenotype in LFS is quite diverse. While specific
TP53 genotype:phenotype correlations have not been clearly demonstrated, several genetic modifier effects are reported. In particular, the mean age of onset of tumors is significantly less in
TP53 mutation carriers who carry an
MDM2 SNP309 G allele compared with those homozygous for the T allele.
58 Similarly, carriers of the
TP53 codon 72 Arginine allele have an earlier tumor onset than those who harbor a homozygous Proline allele.
58 The cumulative combination of
MDM2 SNP309 and
TP53 codon 72 status, telomere length in peripheral blood cells, and possibly specific
TP53 mutations may eventually be used as a predictive biomarker for cancer type and age of onset in LFS.
60 DNA copy number variation (CNV) is strikingly enriched in the constitutional DNA of
TP53 mutation carriers, and these CNVs can be inherited and frequently encompass other cancer genes, suggesting that the genomic instability conferred by the
TP53 mutation can be transmitted from generation to generation.
61 The potential role of telomere attrition and aberrant CNVs is exemplified by evidence that demonstrates chromothripsis (chromosome shattering and reshuffling) in tumors, such as medulloblastomas, from patients harboring germline
TP53 mutations.
62A large number of studies have analyzed groups of patients with tumors characteristic of LFS, yet lacking characteristic family histories of cancer, for germline
TP53 mutations. A few examples are provided here. For example, mutations have been identified in approximately 50% of children with ACC,
63 10% of children with osteosarcoma,
64 and 10% of children with rhabdomyosarcoma.
65 In the latter study, the age average of onset (22 months) was lower for
TP53 mutation carriers. Strikingly, 75% of children with anaplastic RMS harbor germline TP53 mutations.
66 Furthermore, one-third of children with sarcomas plus either multiple primary tumors, or a significant family history of cancer have germline
TP53 mutations.
67 Studies reveal that more than 90% of ACC patients from Brazil carry the
TP53 R337H missense mutation as a founder mutation.
68 ACC diagnoses are overrepresented in families carrying this mutation; however, many Brazilian families exhibit the classic LFS phenotype of multiple cancer types.
69 Evidence that germline
TP53 mutations are associated with specific subtypes of childhood cancer was further validated by the recent finding that apparently germline
TP53 mutations were identified in 50% of children with the rare hypodiploid form of ALL.
70Presymptomatic molecular testing for
TP53 germline mutations in members of LFS kindreds has been met with significant controversy. A major step forward came with the report of early tumor detection with favorable impact on survival in a small cohort of childhood and adult
TP53 mutation carriers screened using a comprehensive tumor surveillance protocol.
71 This protocol (often referred to as the “Toronto Protocol”) uses a combination of annual rapid sequence whole-body magnetic resonance imaging (MRI), dedicated brain and breast (for young adult) MRI, and frequent abdominal/pelvic ultrasounds and several biochemical assays in at-risk individuals (see
Table 2.4). Relatively long-term follow-up has demonstrated that the approach is feasible and offers a significant survival advantage and reduced treatment morbidity when compared with carriers who do not undergo surveillance. The Toronto protocol does not include PET-CT evaluation, although a few anecdotal reports of detection of ACC with PET-CT imaging have been published in children with LFS.
72,73 With the implementation of the Toronto protocol, many additional centers are beginning presymptomatic testing of children at risk for
TP53 mutations. Clinical evaluation of children at increased genetic risk for cancer is discussed further in the last section of this chapter.