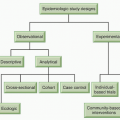
As early as the 1800s, initial observations of unusual cancer incidences in occupational groups provided the first indications that chemicals were a cause of human cancer, which was then confirmed in experimental animal studies during the early and mid 1900s. However, the extent to which chemical exposures contribute to cancer incidence was not fully appreciated until population-based studies documented differing organ-specific cancer rates in geographically distinct populations and in cohort studies such as those that linked smoking to lung cancer.1 The most commonly occurring chemical exposures that increase cancer risk are tobacco, alcoholic beverages, diet, and reproductive factors (e.g., hormones). Today, it is recognized that cancer results not solely from chemical exposure (e.g., in the workplace or at home), but that a variety of biologic, social, and physical factors contribute to cancer pathogenesis.2,3 For some common cancers, it also has been recognized that heritable factors also contribute to cancer risk from chemical exposure (e.g., genes involved in carcinogen metabolism, DNA repair, a variety of cancer pathways).4 Twin studies show that for common cancers, nongenetic risk factors are dominant, and the best associations for genetic risks of sporadic cancers indicate that the risks for specific genetic traits are typically less than 1.5-fold.5,6,7 The role of the tumor microenvironment, the cancer stem cells, and feedback signaling to and from the tumor also have been recently recognized as important contributors to carcinogenesis, although how chemicals affect these have yet been clearly demonstrated.8,9,10
The experimental induction of tumors in animals, the neoplastic transformation of cultured cells by chemicals, and the molecular analysis of human tumors have revealed important concepts regarding the pathogenesis of cancer and how laboratory studies can be used to better understand human cancer pathogenesis.7,11,12 Chemical carcinogens usually affect specific organs, targeting the epithelial cells (or other susceptible cells within an organ) and causing genetic damage (genotoxic) or epigenetic effects regulating DNA transcription and translation. Chemically related DNA damage and consequent somatic mutations relevant to human cancer can occur either directly from exogenous exposures or indirectly by activation of endogenous mutagenic pathways (e.g., nitric oxide, oxyradicals).13,14 The risk of developing a chemically induced tumor may be modified by nongenotoxic exogenous and endogenous exposures and factors (e.g., hormones, immunosuppression triggered by the tumor), and by accumulated exposure to the same or different genotoxic carcinogens.7,15
Analyses of how chemicals induce cancer in animal models and human populations has had a major impact on human health. Experimental studies have been instrumental in replicating hypotheses generated from human studies and identifying pathobiologic mechanisms. For example, animal experiments confirmed the carcinogenic and cocarcinogenic properties of cigarette smoke and identified bioactive chemical and gaseous components.1 The transplacental carcinogenicity of diethylstilbestrol and the hazards of specific occupational carcinogens such as vinyl chloride, benzene, aromatic amines, and bis(chloromethyl)ether led to a reduction in allowable exposures of suspected human carcinogens from the workplace and a reduction in cancer rates. Dietary factors that enhance or inhibit cancer development and the contribution of obesity to specific organ sites have been identified in models of chemical carcinogenesis, and alterations in diet and obesity are expected to result in reduced cancer risk. Experimental animal studies are the mainstay of risk assessment as a screening tool to identify potential carcinogens in the workplace and the environment, although these studies do not prove specific chemical etiologies as a cause of human cancer because of interspecies differences and the use of maximally tolerated doses that do not replicate human exposure.
THE NATURE OF CHEMICAL CARCINOGENS: CHEMISTRY AND METABOLISM
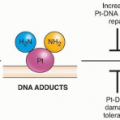
The National Toxicology Program, based mostly on experimental animal studies and supported by epidemiology studies when available, lists 45 chemical, physical, and infectious agents as known human carcinogens and about 175 that are reasonably anticipated to be human carcinogens (http://ntp.niehs.nih.gov/?objectid=035E57E7-BDD9-2D9B-AFB9D1CADC8 D09C1), whereas the International Agency for Research on Cancer (IARC) lists 113 agents as carcinogenic to humans and 66 that are probably carcinogenic to humans (http://monographs.iarc.fr/ENG/Classification/index.php). Table 7.1 provides a selected list of known human carcinogens, as indicated by the IARC, which are continuously updated.16 Most chemical carcinogens first undergo metabolic activation by cytochrome P450s or other metabolic pathways so that they react with DNA and/or alter epigenetic mechanisms.11,17 This process, evolutionarily presumed to have been developed to rid the body of foreign chemicals for excretion, inadvertently generates reactive carcinogenic intermediates that can bind cellular molecules, including DNA, and cause mutations or other alterations.18 Recent data indicate that metabolizing enzymes also have the ability to cross-talk with transcription factors involved in the regulation of other metabolizing and antioxidant enzymes.19 DNA is considered the ultimate target for most carcinogens to cause either mutations or gross chromosomal changes, but epigenetic effects, such as altered DNA methylation and gene transcription, also promote carcinogenesis.20 The formation of DNA adducts, where chemicals bind directly to DNA to promote mutations, is likely necessary but not sufficient to cause cancer.
Genotoxic carcinogens may transfer simple alkyl or complexed (aryl) alkyl groups to specific sites on DNA bases.18,21 These alkylating and aryl-alkylating agents include, but are not limited to, N-nitroso compounds, aliphatic epoxides, aflatoxins, mustards, polycyclic aromatic hydrocarbons, and other combustion products of fossil fuels and vegetable matter. Others transfer arylamine residues to DNA, as exemplified by aryl aromatic amines, aminoazo dyes, and heterocyclic aromatic amines. For genotoxic carcinogens, the interaction with DNA is not random, and each class of agents reacts selectively with purine and pyrimidine targets.7,18,21 Furthermore, targeting carcinogens to particular sites in DNA is determined by nucleotide sequence, by host cell, and by selective DNA repair processes (see later discussion), making some genetic material at risk over others. As expected from this chemistry, genotoxic carcinogens can be potent mutagens and particularly adept at causing nucleotide base mispairing or small deletions, leading to missense or nonsense mutations. Others may cause macrogenetic damage, such as chromosome breaks and large deletions. In some cases, such genotoxic damage may result in changes in transcription and translation that affect protein levels or function, which in turn alter the behavior of the specific host cell type. For example, there may be effects on cell proliferation, programmed cell death, or DNA repair. This is best typified by the signature mutations detected in the p53 gene caused by ingested aflatoxin in human liver cancer22 and by polycyclic aromatic hydrocarbons human lung cancer caused by the inhalation of cigarette smoke.15,23,24 Similarly, a distinct pattern of mutations is detected in pancreatic cancers from smokers when compared with pancreatic cancers from nonsmokers.25
Magenta manufacturing, auramine manufacturing, painting, rubber production
Transitional cell cancer
Prostate
Cadmium
–
Adenocarcinoma
Skin
Arsenic, benzo(a)pyrene, coal tar and pitch, mineral oils, soot, cyclosporin A, azathioprine, shale oils
–
Squamous cell cancer, basal cell cancer
Bone marrow
Benzene, tobacco smoke, ethylene oxide, antineoplastic agents, cyclosporin A, formaldehyde
Rubber workers
Leukemia, lymphoma
aThe carcinogen designations are determined by the International Agency for Research on Cancer (http://monographs.iarc.fr/index.php). They do not imply proof of carcinogenicity in individuals. This table is not all inclusive. For additional information, the reader is referred to agency documents and publications.
Some chemicals that cause cancers in laboratory rodents are not demonstrably genotoxic. In general, these agents are carcinogenic in laboratory animals at high doses and require prolonged exposure. Synthetic pesticides and herbicides fall within this group, as do a number of natural products that are ingested. The mechanism of action by nongenotoxic carcinogens is not well understood, and may be related in some cases to toxic cell death and regenerative hyperplasia. They may also induce endogenous mutagenic mechanisms through the production of free radicals, increasing rates of depurination, and the deamination of 5-methylcytosine. In other cases, nongenotoxic carcinogens may have hormonal effects on hormone-dependent tissues. For example, some pesticides, herbicides, and fungicides have endocrine-disrupting properties in experimental models, although the relation to human cancer risk is unknown.
ANIMAL MODEL SYSTEMS AND CHEMICAL CARCINOGENESIS
Most human chemical carcinogens can induce tumors in experimental animals; however, the tumors may not be in the same organ, the exposure pathways may differ from human exposure, and the causative mechanisms may not exist in humans. In many cases, however, the cell of origin, morphogenesis, phenotypic markers, and genetic alterations are qualitatively identical to corresponding human cancers. Furthermore, animal models have revealed the constancy of carcinogen-host interaction among mammalian species by reproducing organ-specific cancers in animals with chemicals identified as human carcinogens, such as coal tar and squamous cell carcinomas, vinyl chloride and hepatic angiosarcomas, aflatoxin and hepatocellular carcinoma, and aromatic amines and bladder cancer. The introduction of genetically modified mice designed to reproduce specific human cancer syndromes and precancer models has accelerated both the understanding of the contributions of chemicals to cancer causation and the identification of potential exogenous carcinogens.26,27 Furthermore, construction of mouse strains genetically altered to express human drug-metabolizing enzymes has added both to the relevance of mouse studies for understanding human carcinogen metabolism and the prediction of genotoxicity from suspected human carcinogens and other chemical exposures.28 Together, these studies have indicated that carcinogenic agents can directly activate oncogenes, inactivate tumor suppressor genes, and cause the genomic changes that are associated with autonomous growth, enhanced survival, and modified gene expression profiles that are required for the malignant phenotype.29
Genetic Susceptibility to Chemical Carcinogenesis in Experimental Animal Models
The use of inbred strains of rodents and spontaneous or genetically modified mutant strains have led to the identification and characterization of genes that modify risks for cancer development.30,31,32 For a variety of tissue sites, including the lungs, the liver, the breast, and the skin, pairs of inbred mice can differ by 100-fold in the risk for tumor development after carcinogen exposure. Genetically determined differences in the affinity for the aryl hydrocarbon hydroxylase (Ah) receptor or other differences in metabolic processing of carcinogens is one modifier that has a major impact on experimental and presumed human cancer risk.33,34,35 The development of mice reconstituted with components of the human carcinogen-metabolizing genome should facilitate the extrapolation of metabolic activity by human enzymes and cancer risk.27,28,36 Such mice also show that other loci regulate the growth of premalignant foci, the response to tumor promoters, the immune response to metastatic cells, and the basal proliferation rate of target cells.30 In mice susceptible to colon cancer due to a carcinogen-induced constitutive mutation in the APC gene, a locus on mouse chromosome 4 confers resistance to colon cancer.31 The identification of the phospholipase A2 gene at this locus and subsequent functional testing in transgenic mice revealed an interesting paracrine protective influence on tumor development.31 This gene, and several other genes mapped for susceptibility to chemically induced mouse tumors (PTPRJ, a receptor type tyrosine phosphatase, and STK6/STK15, an aurora kinase), have now been shown to influence susceptibility to organ-specific cancer induction in humans.30,31
Only gold members can continue reading. Log In or Register to continue