Abstract
Bone marrow (BM) hyperplasia, a non-neoplastic expansion of one or more of the haematopoietic cell lineages due to an increased number of cells, can manifest in a range of morphological appearances depending on the underlying cause. Similarly to other tissue types, hyperplasia is often associated with an increase in the number of cells with less mature morphology. It is this reactive atypia/dyshaemopoietic morphology that needs careful assessment and correlation/integration with clinical, biochemical, radiological and often molecular findings to correctly interpret the underlying process and avoid misdiagnosis as a neoplastic proliferation. This chapter will consider erythroid, myeloid and megakaryocytic hyperplasia. Reactive conditions of histiocytes are covered in Chapter 6.
Introduction
Bone marrow (BM) hyperplasia, a non-neoplastic expansion of one or more of the haematopoietic cell lineages due to an increased number of cells, can manifest in a range of morphological appearances depending on the underlying cause. Similarly to other tissue types, hyperplasia is often associated with an increase in the number of cells with less mature morphology. It is this reactive atypia/dyshaemopoietic morphology that needs careful assessment and correlation/integration with clinical, biochemical, radiological and often molecular findings to correctly interpret the underlying process and avoid misdiagnosis as a neoplastic proliferation. This chapter will consider erythroid, myeloid and megakaryocytic hyperplasia. Reactive conditions of histiocytes are covered in Chapter 6.
Erythroid Hyperplasia
Hyperplasia of the erythroid lineage can be the end result of numerous different pathological processes affecting the finely regulated pathway of erythropoiesis (Table 5.1) [1]. Depending on the underlying pathogenetic mechanism, it can either be an isolated phenomenon or be associated with changes affecting the other two BM lineages. As a morphological phenomenon it is either associated with peripheral erythrocytosis or anaemia.
Table 5.1 Causes of erythroid hyperplasia.
| Congenitala | Acquired |
|---|---|
| Primary familial and congenital polycythaemia Haemoglobinopathies | Blood loss Haematinic deficiency Haemolysis Hypoxia (high altitude or other hypoxic states) |
a The detailed discussion of congenital disorders affecting erythrocyte production and haemoglobin production are beyond the scope of this book. Please refer to Diagnostic Pediatric Hematopathology edited by Maria A. Proytcheva, Cambridge University Press 2011.
Isolated erythroid hyperplasia is rare and is more likely to be associated with congenital cases and therefore mostly seen in a paediatric haematopathology practice.
Erythroid hyperplasia in acquired cases is rarely isolated and the other two BM lineages are often affected to variable degree.
Short-term regulation of erythroid production is orchestrated by erythropoietin (EPO), a cytokine produced in the kidneys in response to hypoxic conditions. Increased EPO levels result in the terminal proliferation of colony forming unit-erythroid (CFU-E). Erythropoietin has a limited effect and after exhausting the three to five division cell cycle capacity of CFU-E under chronic stress (such as haemolysis) a number of other additional regulatory factors (SCF, insulin like growth factor 1 (IGF-1), glucocorticoids (GCs), and IL-3, and IL-6) regulate the production of more CFU-E from burst-forming unit-erythroid (BFU-E), the earliest erythroid lineage-specific precursor [2].
Morphology
Histo- and cytomorphological features are the result and end product of various pathological processes, which are almost identical regardless of aetiology. The BM is usually normocellular or hypercellular owing to hyperplasia of the erythroid lineage with a decreased/reversed myeloid to erythroid ratio. In most cases the main structural finding is the expansion of erythroid islands, which often bridge or run together depending on the degree of response. Depending on the chronicity of the process, immature forms such as erythroblasts and proerythroblasts increase in number giving an impression of immature islands. It is important to note that while these enlarged islands might appear immature in each of these there is a range of forms at different levels of maturity, i.e. there is left-shifted but full, sequential maturation. Immaturity and left shift are often associated with, and indeed often called, dysplasia. However, in true dyserythropoiesis there is synchronicity of maturation within and dyssynchrony between islands, i.e. most forms are at the same level of maturation within the islands and at different levels when compared to a neighbouring island. There is no endosteal translocation of erythroid islands, although it may appear so purely for positional reason and due to the degree of expansion.
In aspirates one can appreciate the expansion of erythropoiesis and reversal of the myeloid to erythroid ratio in addition to mild dysmorphic/dyspoietic features, such as nuclear-cytoplasmic dyssynchrony, ragged cytoplasm and poor cytoplasmic haemoglobinization. However, these are present in less than 10% of erythroid lineage cells. When an isolated finding, one must consider the possibility of early/emerging myelodysplastic syndrome with unilineage dysplasia. Therefore correlation with an iron stain, histomorphology, cytogenetic and potentially molecular genetic findings is essential in all cases.
Erythroid Hyperplasia Driven by Hypoxia and Increased Erythropoietin Production
Numerous pathogenetic mechanisms may lead to hypoxia and subsequent upregulation of EPO production by the kidneys, including acute/subacute blood loss and hypoxia (heart/lung disease, high altitude or other hypoxic states) [3]. Use of synthetic EPO (medical and doping) will result in a similar morphological picture. Morphological assessment of the BM reveals hyperplasia of the erythroid lineage as described in the ‘Morphology’ section.
Erythroid Hyperplasia Driven by Destruction of Erythrocytes
Extrinsic causes of haemolytic anaemia are summarized in Table 5.2 (intrinsic causes, such as haemoglobinopathies, are beyond the scope of this book). Most of these aetiological factors will cause secondary autoimmune haemolytic anaemia (AIHA) accounting for about half of AIHA cases. Diagnostic work-up necessitates thorough examination of the patient’s medical history, serological findings and direct agglutinin test (DAT). Based on the type of anti-erythrocyte antibody and the thermal optimum of erythrocyte binding used to detect anti–erythrocyte antibodies these disorders can be grouped into warm AIHA (WAIHA, reacts maximally at 37°C) and cold agglutinin disease (CAD, reacts maximally at <4°C). Autoantibodies with IgG isotype are more common in WAIHA and IgM in CAD. IgA isotype autoantibodies are rare and cause severe haemolysis [4].
Table 5.2 Extrinsic/acquired causes of haemolysis.
| Autoimmune haemolytic anaemia – primary/idiopathic | ||
| Autoimmune haemolytic anaemia – secondary | Autoimmune disease | Systemic lupus erythematosus (SLE) Rheumatoid arthritis Ulcerative colitis Scleroderma Crohn’s disease |
| Underlying malignancy | Haematological malignancy Non-haematological malignancy | |
| Infections | Viral (e.g. hepatitis viruses, EBV) Bacterial (e.g. E. coli, streptococci) | |
| Drugs | Acetaminophen Antibiotics (e.g. penicillin, ampicillin, methicillin) Chlorpromazine Ibuprofen Interferon alfa Procainamide Quinine Rifampin | |
| Mechanic | Mechanic heart valve Haemodialysis | |
| Transfusion reaction |
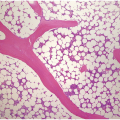
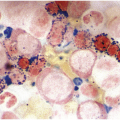
Bone marrow examination is usually performed to confirm/exclude underlying haematological or non-haematological malignancy. The marrow itself is usually hypercellular with erythroid hyperplasia (Figure 5.1). When the aetiological factor is neoplastic it might be identified and further characterized by morphological and immunohistochemical assessment. Autoimmune diseases are often associated with mild expansion of polytypic plasma cells. In infective settings the infectious agent, such as EBV, might be readily identifiable in the marrow and other lineages can also be affected and show reactive dyshaemopoietic changes.
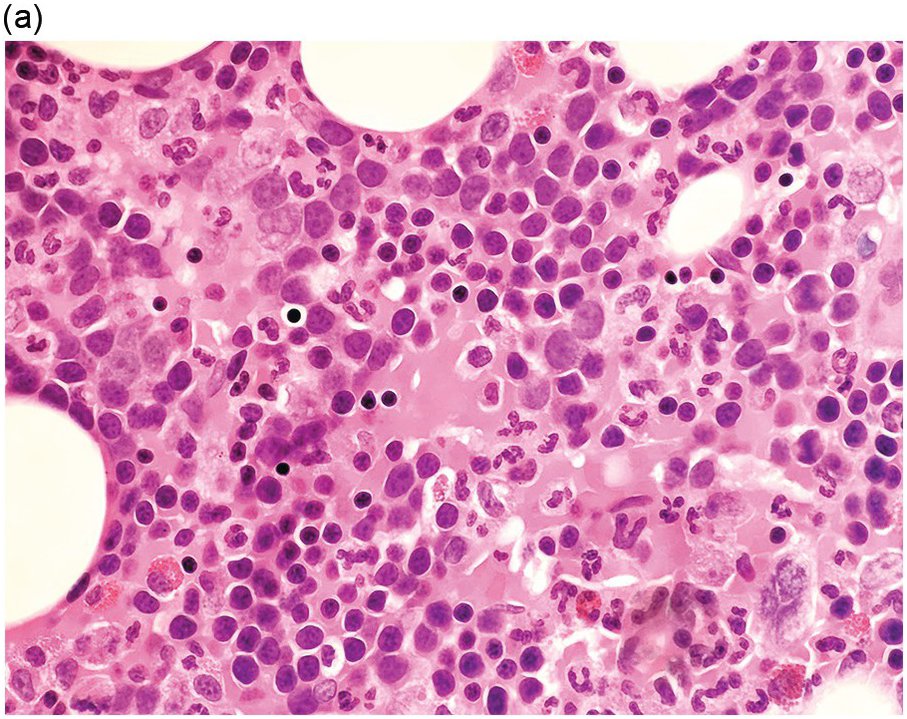
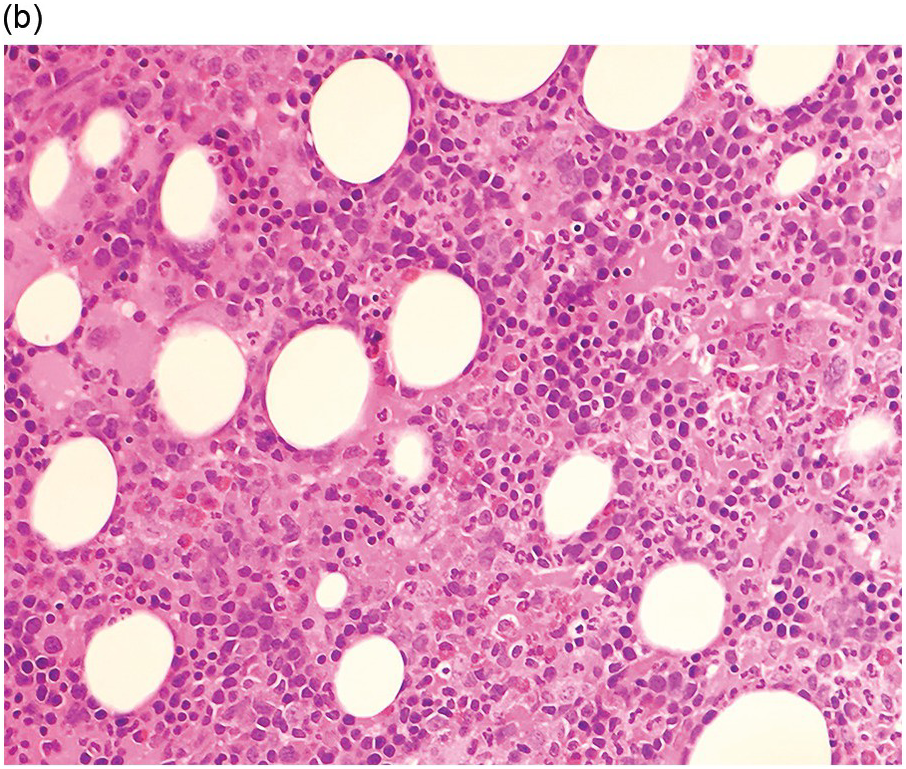
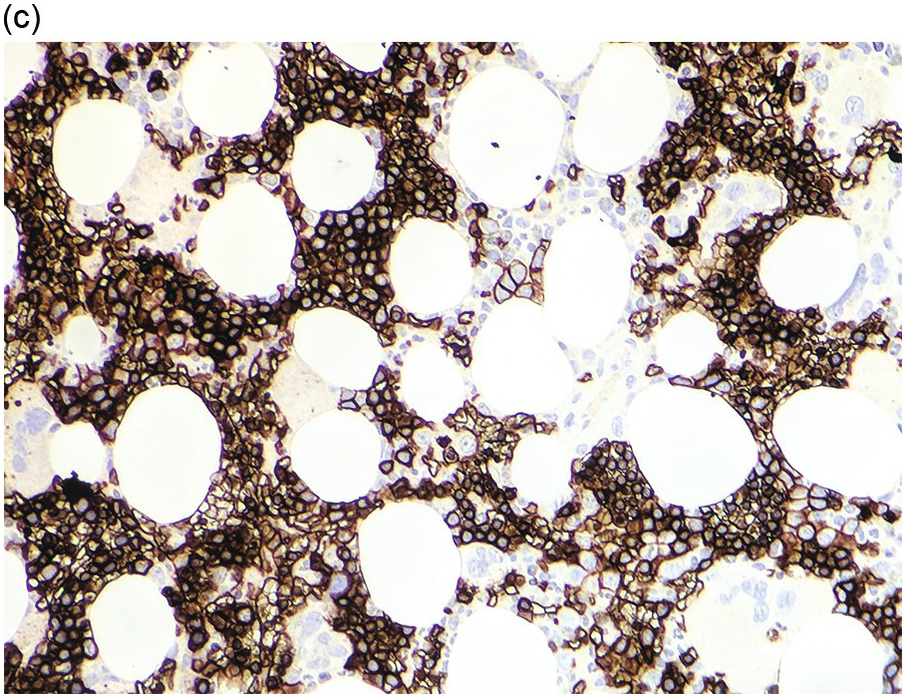
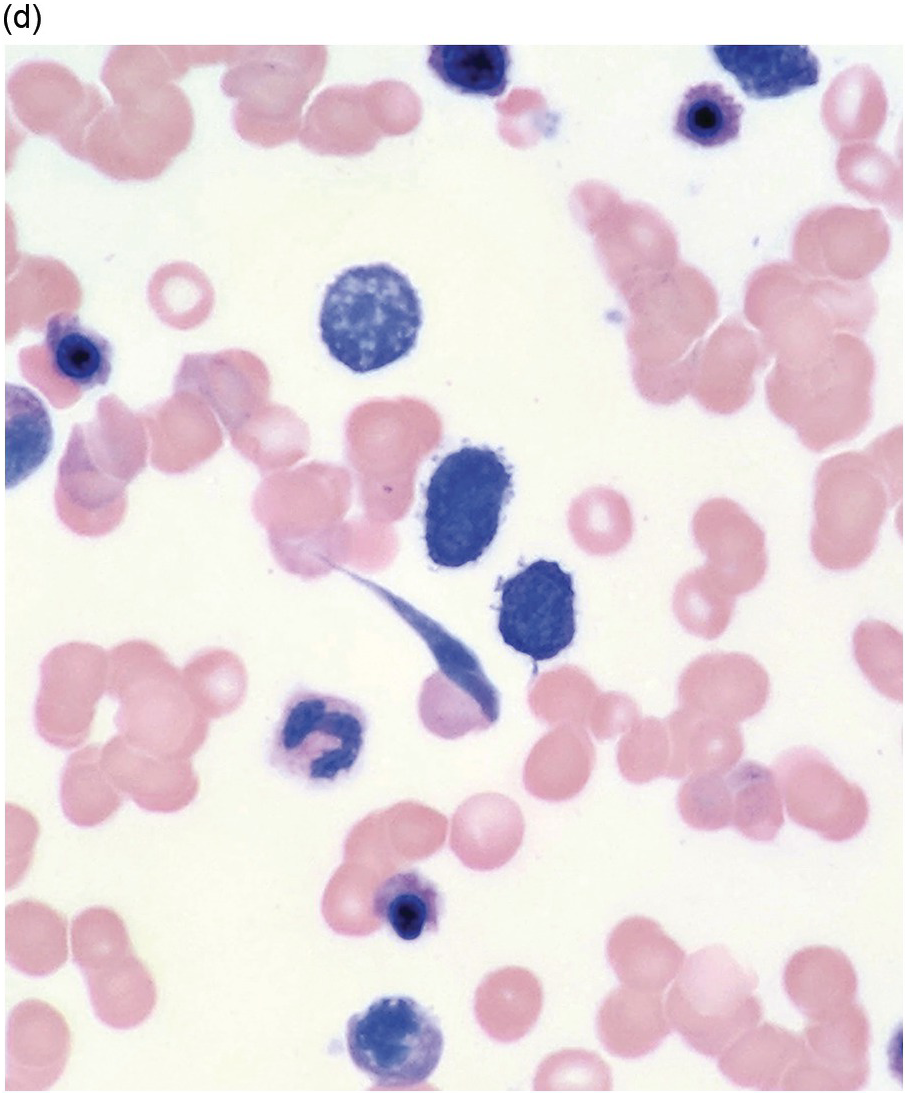
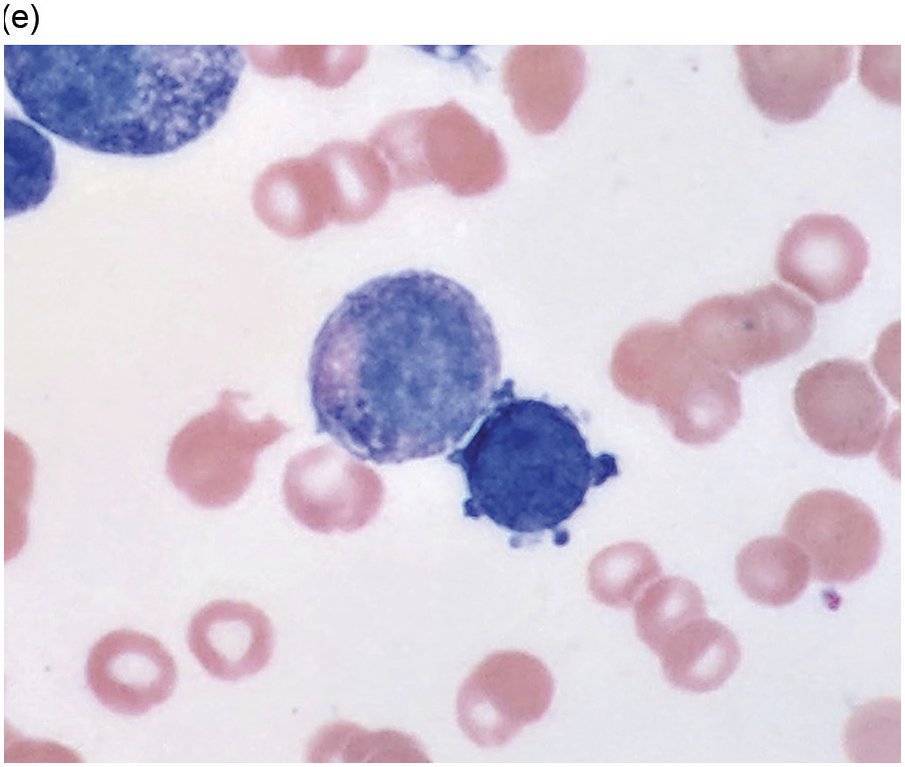
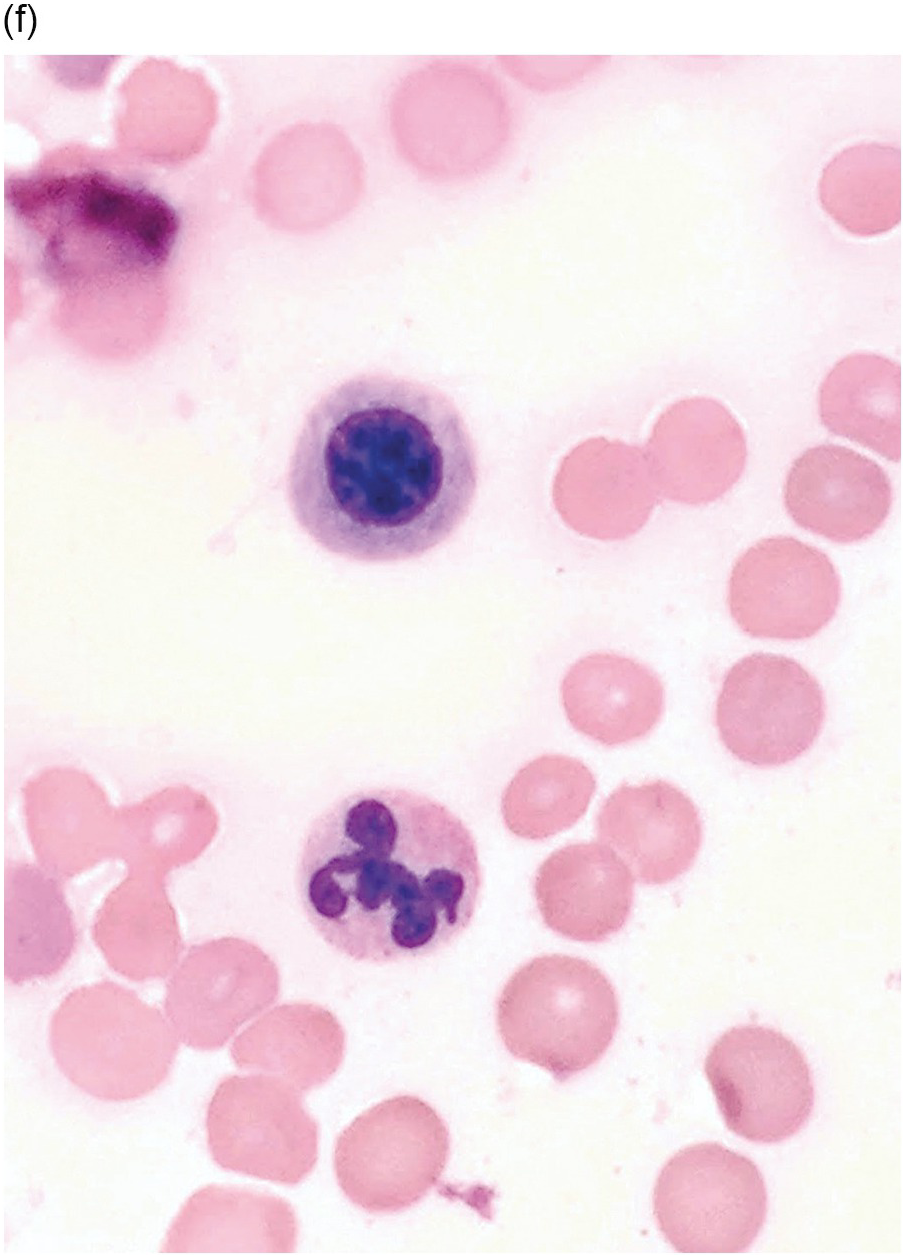
Figure 5.1 Bone marrow morphology in a 67-year-old patient with haemolytic anaemia. Erythroid hyperplasia is evident on the haematoxylin and eosin (H&E)-stained section (a and b). Erythroid islands are enlarged and run together with cells at various stages of maturation (a). The same features can be seen on sections stained with anti-glycophorin A antibody (c). Mild dyshaemopoietic changes in the aspirate sample: ragged cytoplasm (d and e) and nuclear-cytoplasmic dyssynchrony (f).
Erythroid Hyperplasia due to Haematinic Deficiency
Megaloblastic Anaemia
Aetiology and Clinical Features. Megaloblastic anaemia is an acquired maturation disorder caused by B12 and folate deficiency, both of which are essential for DNA synthesis. Low levels (Table 5.3) result in an inability to progress from G2 growth stage to the mitosis (M) stage resulting in continuing cell growth and macrocytosis [5].
Table 5.3 Causes of megaloblastic anaemia.
| Diet | Vitamin B12 deficiency | Veganism, poor diet |
| Folate deficiency | Poor-quality diet (old age, poverty) Alcoholism | |
| Malabsorption | Gastric causes of B12 deficiency | Pernicious anaemia Congenital intrinsic factor deficiency or abnormality Gastrectomy |
| Intestinal causes of B12 deficiency | Stagnant loop, ileal resection Crohn’s disease | |
| Intestinal causes of folate deficiency | Coeliac disease Jejunal resection Tropical sprue | |
| Increased turnover | Folate deficiency | Pregnancy Prematurity Chronic haemolytic anaemia Extensive inflammatory/malignant disease |
| Renal loss | Folate deficiency | Congestive cardiac failure Dialysis |
| Drugs | Folate deficiency | Sulphasalazine, anticonvulsants |
| Defects of metabolism | Defects of vitamin B12 metabolism | Nitrous oxide anaesthesia |
| Defects of folate metabolism | Methotrexate treatment | |
| Inherited disorders | Inherited defects of DNA synthesis | Congenital pernicious anaemia /hereditary intrinsic factor deficiency Megaloblastic anaemia 1/Imerslunf-Grasbeck syndrome Trabscobalamin II deficiency |
| Inherited pyrimidine synthesis disorders |
The earliest finding is of macrocytic anaemia (decreased RBC count and haemoglobin concentration with increased mean corpuscular volume) with normal mean corpuscular haemoglobin concentration and reduced reticulocyte count. More severe cases are associated with progressive decrease in platelet and white cell counts with resulting pancytopaenia due to ineffective cell production.
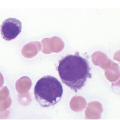
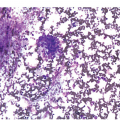
Microscopy. The BM is hypercellular for the age of the patient, predominantly due to erythroid hyperplasia although the other two lineages are also affected (Figure 5.2). Erythroid precursors are prominent and display megaloblastic morphology of increased nuclear size with loose/sieve-like nuclear chromatin despite more mature cytoplasm (nuclear-cytoplasmic dyssynchrony). Although most prominent in erythroid precursors, these features can also be observed in the granulocytic series with giant metamyelocytes, large bands and hypersegmented neutrophils. Megakaryocytes are also enlarged and can be hyperlobated. Karyorrhexis is noticeable throughout.