Abstract
Bone marrow (BM) trephine biopsy (BMB) is a frequent and routine diagnostic investigation, as nicely described in the previous chapters, and is also widely used for follow-up of haematological disorders to judge the effectiveness of therapeutic interventions [1]. In addition, several drugs applied primarily for the treatment of non-haematological disorders may cause serious haematological side effects, such as pancytopaenia, agranulocytosis or anaemia. On occasion, the causative link between the haematological symptoms and the previous drug exposure is not perceived and it is not uncommon for BMB to be obtained in such circumstances without any information of a history of previous drug exposure being made available to the haematopathologist [2].
Introduction
Bone marrow (BM) trephine biopsy (BMB) is a frequent and routine diagnostic investigation, as nicely described in the previous chapters, and is also widely used for follow-up of haematological disorders to judge the effectiveness of therapeutic interventions [1]. In addition, several drugs applied primarily for the treatment of non-haematological disorders may cause serious haematological side effects, such as pancytopaenia, agranulocytosis or anaemia. On occasion, the causative link between the haematological symptoms and the previous drug exposure is not perceived and it is not uncommon for BMB to be obtained in such circumstances without any information of a history of previous drug exposure being made available to the haematopathologist [2].
Above all, drug-induced BM changes are often difficult to interpret and may in some cases mimic malignant conditions [2, 3]. All BM compartments (haematopoiesis, stroma and vessels) are sensitive to exposure to pharmacological agents and the morphological patterns of such responses are largely non-specific, thus giving no direct clue to causality. With the exception of germinal centres, the BM is the most rapidly proliferating tissue in the human body, and regulation of haematopoiesis depends on sufficient presence of ions, especially iron, and metabolites, especially nucleotides, and on the proper action of cytokines and growth factors [4]. Drugs interfering with any of these processes inevitably cause morphological BM changes (Table 18.1 summarizes some typical BM changes following therapy). In particular, arylamine-containing drugs, such as clozapine for example, are oxidized by the myeloperoxidase of granulocytes and precursors to form free radicals that destroy the latter, and can cause neutropaenia or agranulocytosis in patients with an unfavourable enzymogenetic or immunogenetic constellation [5]. Finally, the BM niche is prone to immune deregulation disturbances such as pharmaco-allergic reactions, most probably due to its local immune regulation peculiarities [5–7].
Table 18.1 Bone marrow changes related to drug exposure.
| Drug class | Expected changes |
|---|---|
| Chemotherapy | Anaemia, leukopaenia, thrombopaenia, pancytopaenia All types of changes listed in Table 18.2 Therapy-associated myeloid neoplasms |
| Haematopoietic growth factors and thrombopoietin receptor agonists | Hypercellularity Myelofibrosis MPN-like/CMML-like changes |
| Interferons | Granulomas |
| Antibodies targeting molecules on lymphocytes and plasma cells | Loss of the expression of the respective molecule Neutropaenia (rituximab, brentuximab) Lymphopaenia (alemtuzumab) Anaemia (natalizumab) |
| Azathioprine Mercaptopurine Thioguianine ±Allopurinol (this drug increases the half-life of the above listed and, thus, the probability of adverse effects) | Anaemia, leukopaenia, pancytopaenia Dysmegakaryopoiesis Dyserythropoiesis Megaloblastic changes Myeloid nuclear segmentation abnormalities Iatrogenic lymphoproliferative disorders |
| Drugs listed in Table 18.3 Chloramphenicol | Idiosyncratic reactions: – neutropaenia/agranulocytosis – aplastic anaemia – haemolytic anaemia – DRESS Myeloid hypoplasia/maturation arrest CD4-predominant lymphocytosis Lipogranulomas |
| Antibodies targeting glycoprotein-IIb/IIIa-receptors | Immune thrombocytopaenia Respective BM changes |
| Steroids | Neutrophilia Hypercellularity Myeloid left-shift |
| mTOR-inhibitors | Thrombopaenia Megakaryocytic hyperplasia and atypia |
| Methotrexate (low dose) | Anaemia, leukopaenia, pancytopaenia Aplastic anaemia Atypia in all haematopoietic lineages Iatrogenic lymphoproliferative disorders |
| Antibodies targeting TNF-α | Thrombopaenia, neutropaenia Aplastic anaemia Mycobacteriosis Iatrogenic lymphoproliferative disorders |
This chapter summarizes and illustrates the most common morphological drug-induced changes in the BM by drug classes and adverse reactions, as well as a condition called drug rash with eosinophilia and systemic symptoms (DRESS), with a special emphasis on diagnostic and differential diagnostic clues. In addition, it addresses definitions of treatment response in common haematological malignancies and describes effects of immunosuppressive drugs, some constitutional immunodeficiencies and human immunodeficiency virus (HIV)-myelopathy. Finally, it refers to changes related to haematopoietic cell and BM transplantation.
Bone Marrow Changes Following Therapy
Chemotherapy
Chemotherapy, particularly multimodal chemotherapy, with or without irradiation, is the treatment of choice for many neoplastic diseases. It can be palliative or curative in its intention and aims at either myeloablation or, failing that, effective cytoreduction of the neoplastic haematopoietic clone in cases of primary BM neoplasms, or at the eradication of a non-haematopoietic neoplasm. Systemic chemotherapy has effects on both neoplastic and normal haematopoietic cells, and there is some evidence that the latter manage to repair the respective damage and recover more rapidly, while the former are damaged more severely due to a general perturbation of many intracellular repair mechanisms in cancer [8], and will thus be at least partly eradicated if resistance mechanisms do not evolve. The common BM changes after chemotherapy, especially myeloablative therapy, are summarized in Table 18.2 and Figure 18.1a–d. These can to a large part be explained by increased cell death due to the cytotoxic drugs.
Table 18.2 Bone marrow changes after myeloablative therapy.
| Decreased cellularity to bone marrow aplasia |
| Fibrinoid necrosis |
| Increased bone marrow macrophages ± granulomas |
| Perivascular plasmacytosis |
| Reduction of marrow fat |
| Reticulin fibrosis |
| Siderosis |
| Single cell death |
| Sinus dilatation |
| Stromal oedema |
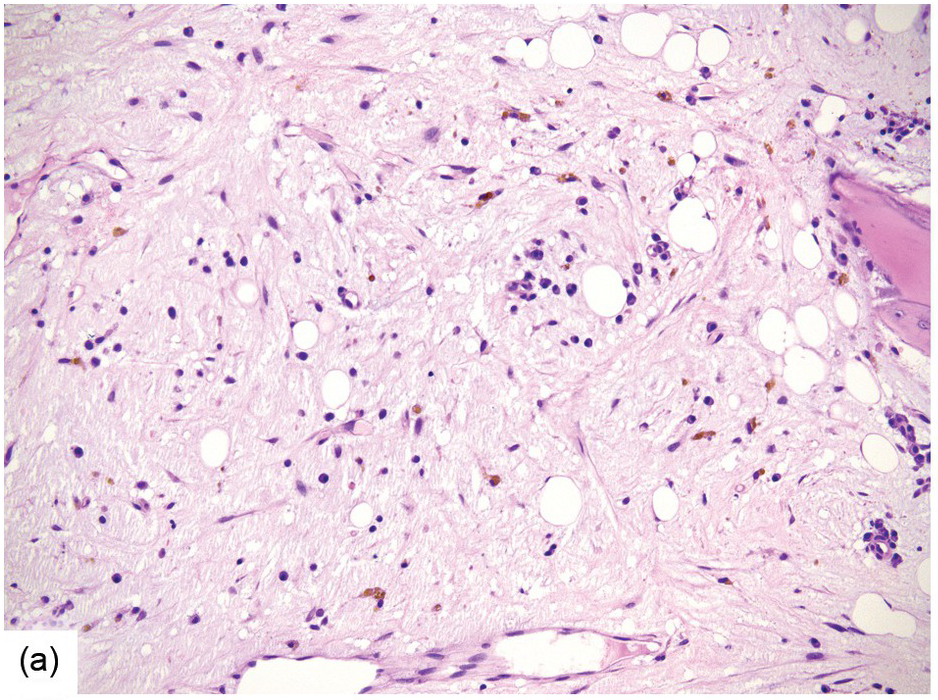
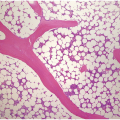
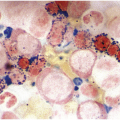
(a) Serous oedema following myeloablative chemotherapy.
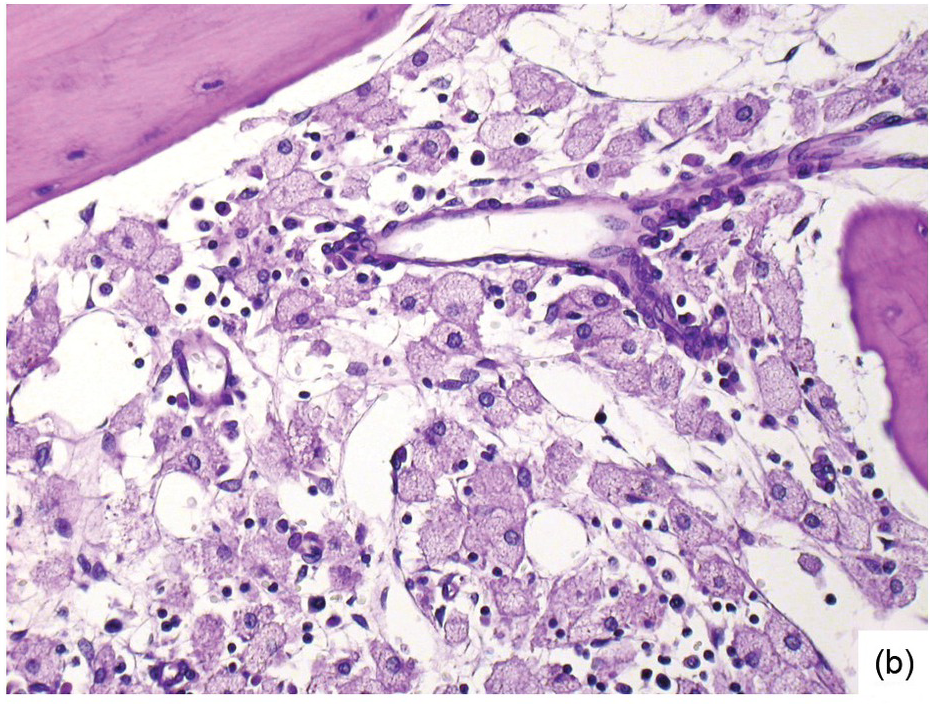
(b) Resorptive post-myeloablative foamy cell reaction in the bone marrow of a respectively treated patient, who suffered from acute myelogenous leukaemia.
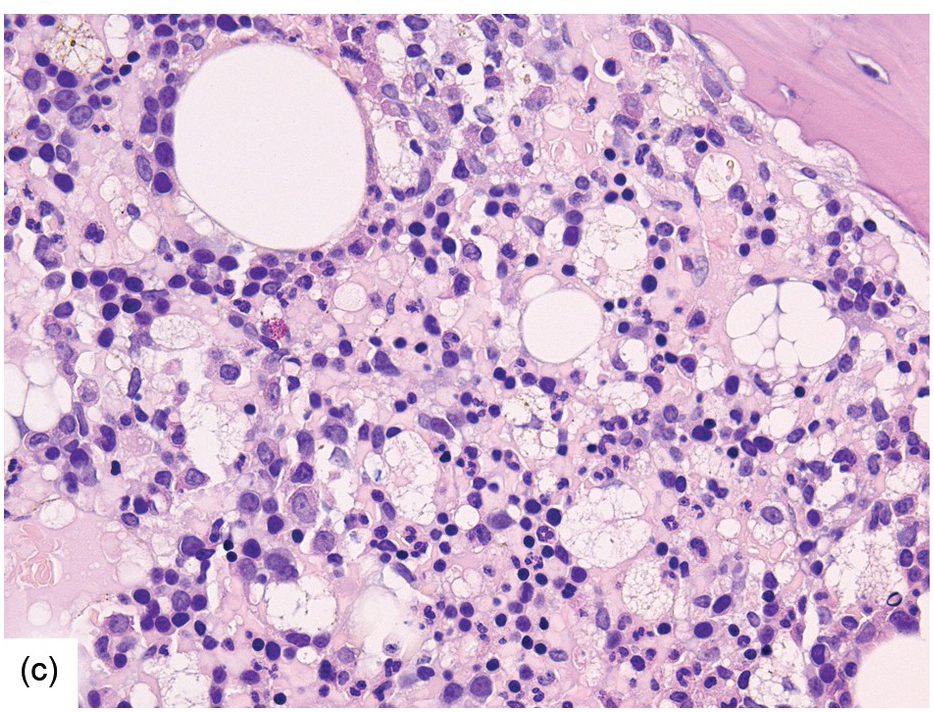
(c) Mixed resorptive and regeneratory changes of the bone marrow following chemotherapy; note the synchronized erythropoiesis and the presence of foamy and haemophagocyting histiocytes.
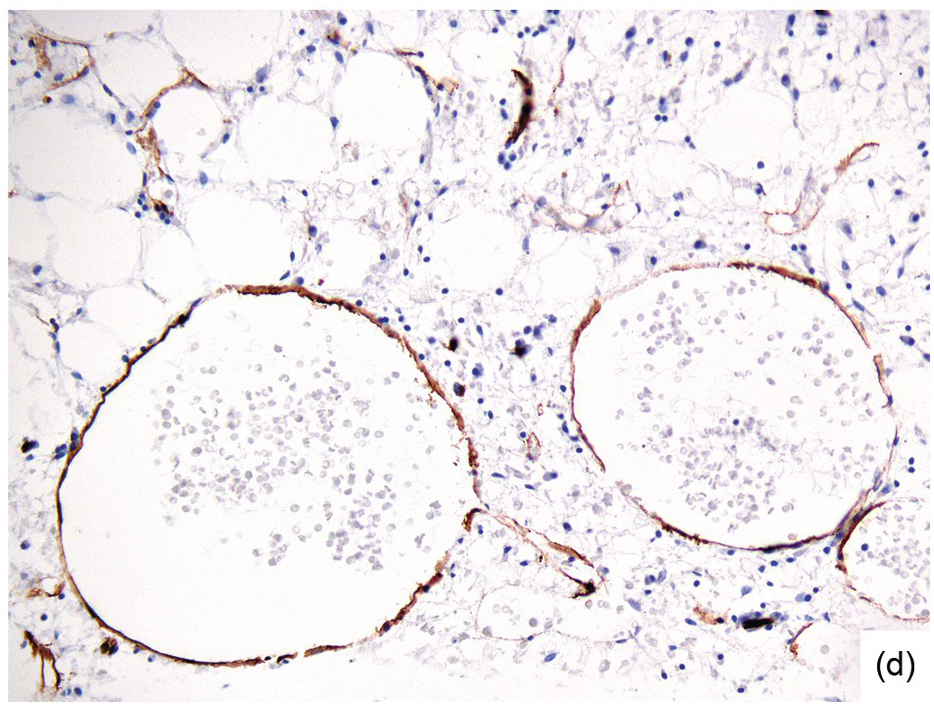
(d) Sinus dilatation after myeloablative therapy (CD34).
In conditions requiring myeloablative therapy, assessment of the treatment response is crucial for further strategic planning, therefore BMB are regularly obtained on days 14–17, 30 and 60 after treatment [9], and haematopathologists will thus face all of the above changes; if reticulin fibrosis is present, BMB might be the only specimen available to assess response. Compared to other methods, BMB has a high sensitivity and specificity for determination of early treatment effects such as overall cellularity, extent of myeloablation and, in particular, non-aspirable foci of residual disease (Figure 18.2), as well as assessment of early regeneration. Along the timeline of the follow-up, BMB after myeloablative therapy will show signs of BM recovery within a week after the last treatment regimen. These include reappearance of fat, recovery with synchronization of the erythroid (typical erythroid colonies) and myeloid lineages (sheets of promyelocytes), the latter being typically peritrabecular in contrast to the perivascular pattern post-transplant. Especially in children, or after T-cell depleted haematopoietic cell transplantation, an increase of haematogones is seen (see below). Subsequently, small megakaryocytes appear in clusters and reticulin fibrosis resolves. Normal age-adjusted BM cellularity is typically reached about four to five weeks after completion of chemotherapy [3, 10].

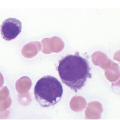
A challenge for the diagnostic haematopathologist is distinguishing reactive (‘regenerative’) atypia from persistent myelodysplasia in early follow-up BMB of patients suffering from myeloid disorders, especially myelodysplastic syndromes (MDS), myelodysplastic/myeloproliferative neoplasms (MDS/MPN) or acute myelogenous leukaemias (AML) [1, 10]. Groups (clusters ≥3 cells) or sheets of blasts with non-peritrabecular distribution, so-called atypically located immature precursors (ALIP), are indicative of persistent neoplasia. Positive CD34 staining is the most useful marker in the distinction of blasts. Megaloblastic changes, dyssynchrony, multinucleation and karyorrhexis of erythropoiesis and pyknotic megakaryocytes are common in the early post-treatment phase, and can be considered as reactive especially following 5-azacitidine-, methotrexate- or mercaptopurine-maintenance chemotherapy (Figure 18.3) [11]. The presence of unequivocal severe dysmegakaryopoiesis, especially micromegakaryocytes, is a strong hint for residual neoplasia, if already present at diagnosis and persisting for more than three months after the last chemotherapeutic cycle (Figure 18.4). Detection of cells bearing recurrent leukaemia-defining aberrations such as PML-RARA or BCR-ABL1 fusions [3] or expression of mutant cytoplasmic NPM1, which can be detected histomorphologically by FISH or immunohistochemistry, unequivocally indicates persistent neoplasia (Figure 18.5). Finally, on many occasions it will be impossible to distinguish reactive changes from residual neoplasia on morphological grounds and the final interpretation must be an integrative diagnosis made after considering the results of the aspiration cytology, flow cytometry (FCM), e.g. the Ogata score [12], cytogenetics and molecular genetics. The value of sensitive sequencing approaches to BMB, though feasible for initial diagnosis of myeloid neoplasms [13], has still not been systematically studied in follow-up biopsies.
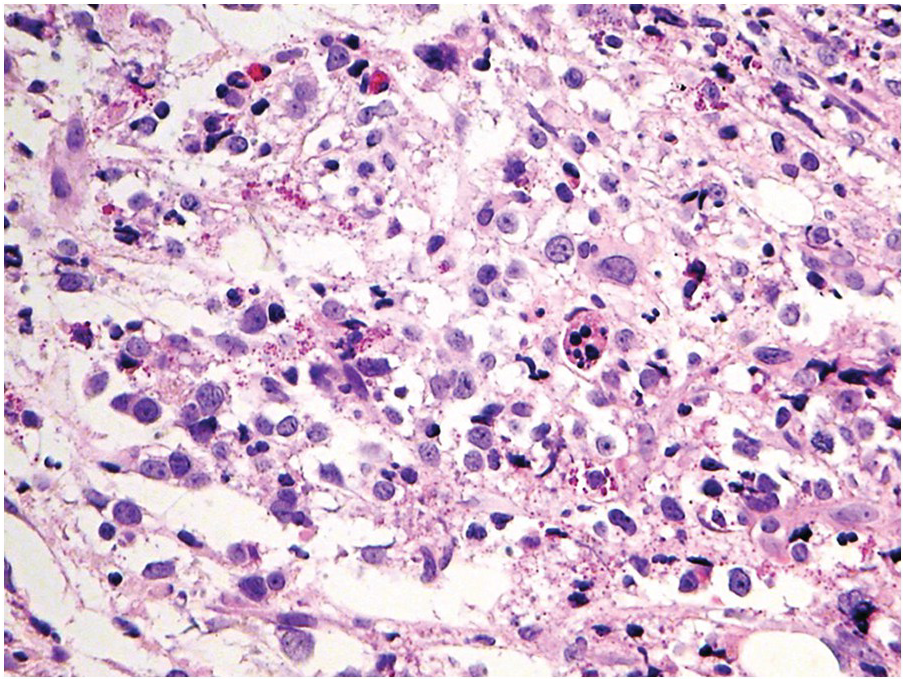
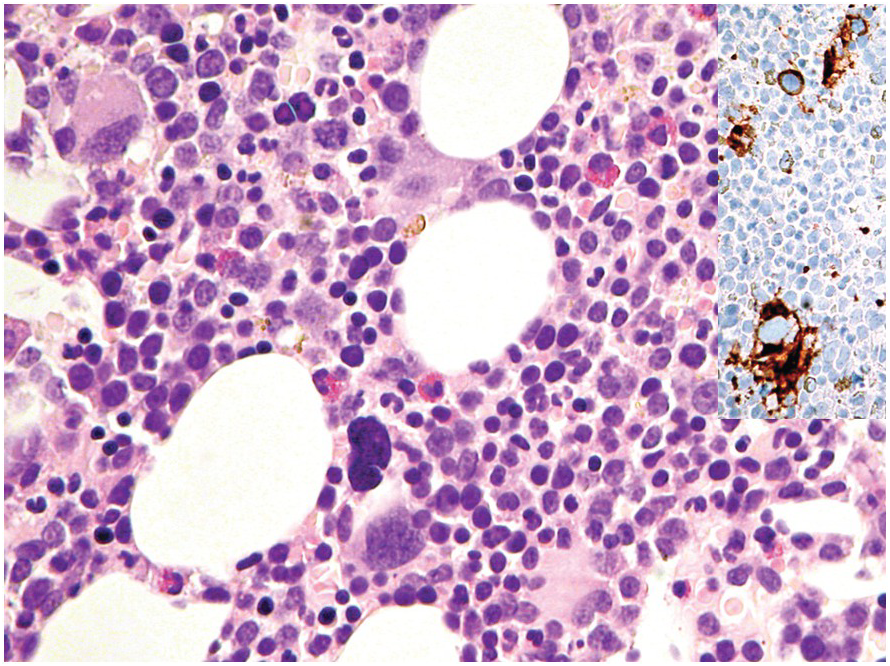
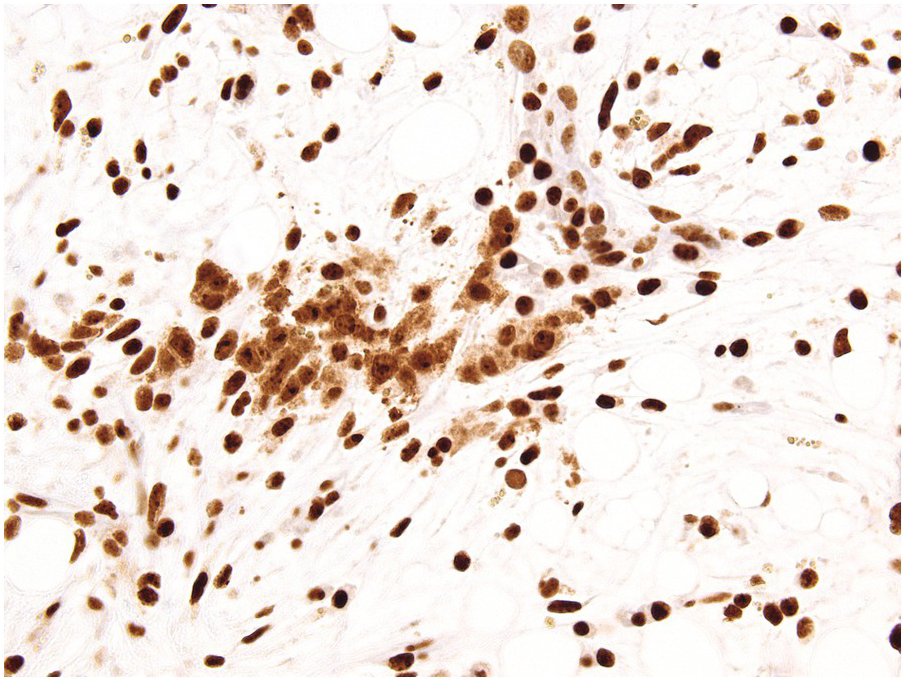
Figure 18.5 Persistent NPM1 mutant blasts on day 17 after myeloablative therapy; note a dozen larger cells with nuclear and cytoplasmic positivity for NPM1 – characteristic of the mutant protein – compared to the sole nuclear positivity of the remaining non-neoplastic cells. Detection of cells bearing recurrent leukaemia-defining aberrations unequivocally indicates persistent neoplasia.
Late effects of cytotoxic therapies, particularly therapy-related myeloid neoplasms, will not be further addressed here and the reader is referred to the relevant book chapters and to excellent reviews [14, 15].
Definition of Response
Evaluation of therapeutic response is, as already mentioned, the most important point in deciding further patient management [9]. Furthermore uniform, clinically relevant and biologically meaningful response criteria, as listed below, and reporting standards, are a prerequisite for comparing any treatment strategy studies. Multiple interdisciplinary methods have to be considered in evaluating response to therapy. Thus, not surprisingly, one should distinguish between clinical, morphologic (cytologic, histologic immunohistochemical), FCM, cytogenetic and molecular responses. Each of these methods has a different sensitivity and specificity in detecting persistent disease. It must be noted that the biological impact of highly sensitive genetic methods in detecting minimal residual disease is sometimes still less clear than anticipated [16] and may require integration of sets/patterns of mutations [17] rather than of single genes, with a few notable exceptions such as NPM1 [18]. From the perspective of the BMB, the sensitivity in detecting minimal residual or early relapsed disease by in situ techniques depends on the unique morphological, phenotypical and genetic features of the neoplastic clone and lies between 0.1 and 10% tumour cell burden [3]; thus, in that respect, BMB is at best an ancillary procedure.
Acute Myelogenous Leukaemia
Clinically relevant recommendations for standard response criteria in AML were published more than a decade ago [19] and, although still valid, are now challenged [20] by considerable changes in treatment strategies, which are applying more targeted agents [21]. Taking into consideration the time and type of prior therapy, peripheral blood cell counts and blast counts, the categories of: (A) early treatment assessment, (B) morphologic leukaemia-free state, (C) morphologic-, cytogenetic- and molecular complete remission, (D) partial remission, and (E) treatment failure, have been introduced. These categories are applicable to BMB and should be used as accurately as possible. A ‘morphologic leukaemia-free state’ is defined as <5% BM blasts, no extramedullary disease and absence of an aberrant leukaemia immunophenotype by FCM during early assessment with persistent neutropaenia (<1 × 109/L) and thrombocytopaenia (<100 × 109/L). A ‘morphologic complete remission’ requires ‘morphologic leukaemia-free’ state as well as an absolute neutrophil count of >1 × 109/L and platelets of >100 × 109/L; there is no requirement for assessment of BM cellularity.
One has to acknowledge that other methods than histopathological assessment are considerably more sensitive in detecting residual disease in AML. Yet in cases of focal blast presence, or in cases with inaspirable blasts, BMB might be the only specimen for response evaluation. Importantly, since detection of persistent disease by any method will have a significant impact on the future treatment strategy decision, the expectations for BMB examination for early treatment response might be very high, despite lack of systematic evidence of the prognostic importance of <5% blasts with unique immunohistochemical or FISH-characteristics in BMB. Examples of relevant situations include core-binding factor AML patients with residual disease, who might benefit from maintenance hypomethylating agent therapy, and various tyrosine kinase inhibitors, which could be viable maintenance options for AML patients with FLT3 or KIT mutations [20]. Likewise, IDH1 or IDH2 inhibitors might be effective in respectively mutant patients, while patients failing to obtain complete remission after induction would be candidates for more intensive therapies [22]. As an example, in my experience, the presence of mutant (cytoplasmic) NPM1 expressing cell groups in BMB (Figure 18.5), even accounting for <5% cells, has always been confirmed by evidence of molecular persistence.
Acute Lymphoblastic Leukaemia
Despite not having been specifically addressed in the literature, most probably because peripheral response to prednisone on day 8 is more relevant, the response criteria in acute lymphoblastic leukaemia (ALL) should be similar to those in AML, i.e. <5% BM blasts, absence of extramedullary disease and neutrophil and platelet recovery, which should be considered to be complete morphologic remission [23]. Usually BMB are obtained on day 15 to assess the percentage of residual blasts. In a recent study, prospective data provided sufficient evidence to categorize BM day 15 responses as M1 (<5%), M2 (5 to <25%) and M3 (≥25% lymphoblasts), allowing better outcome prediction than the peripheral response on day 8 in B-ALL but not in T-ALL [24]. The authors suggest that, while day 8 peripheral response reflects the effect of 7 days of prednisone and one intrathecal dose of methotrexate, day 15 BM findings reflect the response to 14 days of prednisone, one dose of vincristine, daunorubicin and asparaginase, and two doses of intrathecal methotrexate. Thus in B-ALL resistance to prednisone can be counteracted by the other chemotherapeutic drugs, while this seems to be less applicable for T-ALL.
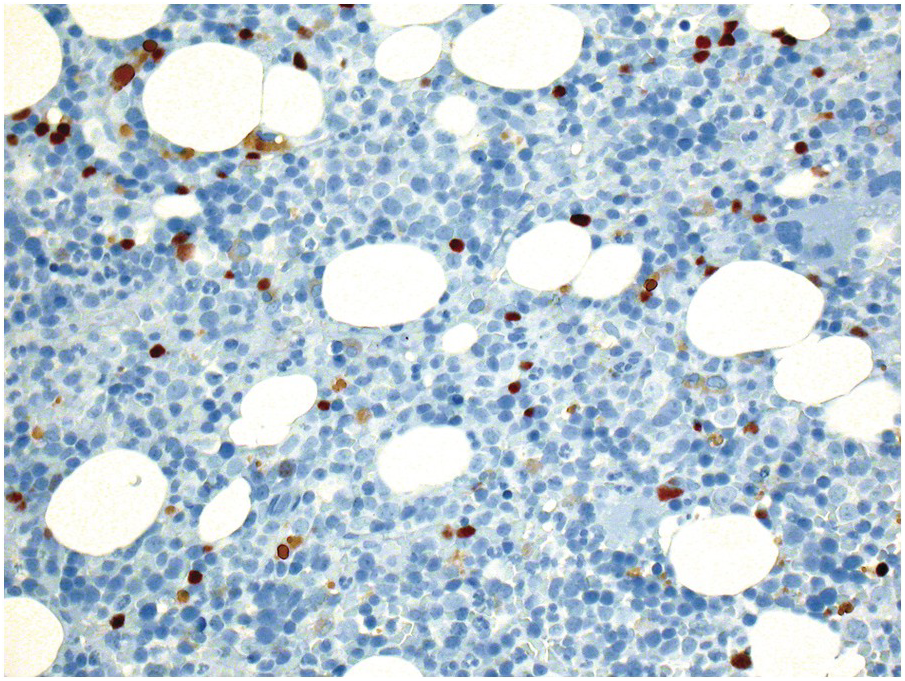
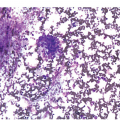
The most important difficulty in early assessment of therapeutic response, particularly in B-ALL, is the distinction between haematogones [25] and persistent malignant lymphoblasts (Figure 18.6). This should be tackled in an integrative way, considering microarchitectural and immunophenotypic features (e.g. CD10, CD19, CD20, CD34, CD79a, CD99 and TdT) as well as by means of FISH in instances bearing recurrent detectable aberration such as e.g. BCR-ABL1 or ETV6-RUNX1 fusions or KMT2A rearrangements. Immunophenotypically haematogone populations exhibit a continuous and complete maturation spectrum of antigen expression characteristic of the normal evolution of B-lineage precursors, lack aberrant or asynchronous antigen expression and rarely form large sheets of ≥7 cells [25, 26]. Thus detection of single cells or small clusters (<7 cells) of immature lymphoid cells with various (i.e. TdT+/CD34+/CD19+/CD10+/CD20–, TdT+/CD34–/CD19+/CD10+/CD20–, TdT+/CD34–/CD19+/CD10+/CD20+ or TdT–/CD34–/CD19+/CD10–/CD20+) but not aberrant or asynchronous phenotypic characteristics, reflecting the sequential marker expression of normal B-cell maturation, and lacking recurrent structural genetic aberrations, if applicable, should be considered consistent with haematogones rather with persistent leukaemic blasts.
Chronic Myelogenous Leukaemia
The majority of chronic myelogenous leukaemia (CML) patients in the chronic phase are now treated with imatinib or other ABL1-tyrosine kinase inhibitors (TKI) [27]. Therapy is followed by clinical (haematologic), morphologic, cytogenetic and molecular remission in most patients and about half achieve a functional cure even after discontinuation of treatment [28]. In a recommendation for uniform response criteria for CML treated with imatinib, assessment of BMB has not been considered mandatory to define treatment response [29]. Nevertheless, the value of follow-up BMB in CML is still essential for detection of focal blasts, accompanying coincidental malignancies, which may be masked at initial presentation (e.g. systemic mastocytosis, Figure 18.7) or for treatment response determination if reticulin fibrosis is present. Therefore knowledge of TKI-induced morphological BM changes, particularly the gradual achievement of CML remission, is important. Typically, TKI induce a prompt reduction in the overall BM cellularity and normalize the myeloid:erythroid ratio. Later on, there is a normalization of megakaryocyte numbers and, finally, of megakaryocyte morphology. If present, BM fibrosis gradually decreases. It has been shown that the presence of >10% abnormal, so-called dwarf megakaryocytes, increased cellularity, myeloid:erythroid ratio >4 and myelofibrosis grade >2 significantly correlate with failure to achieve complete cytogenetic response. Considering these morphologic parameters and three cytological parameters on the corresponding aspirates (dry tap, >5% blasts, >1% basophils), each parameter adding one point, a morphological response score for follow-up BMB and aspirates of TKI-treated patients has been proposed [30]. Indeed not a single patient with a score >2 achieved a complete cytogenetic response.
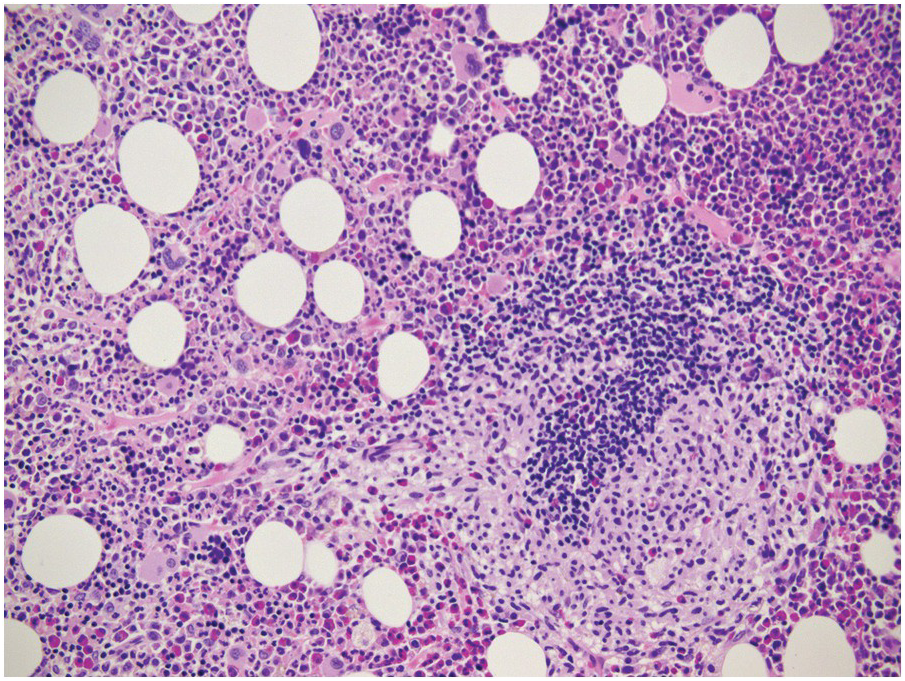
Figure 18.7 Systemic mastocytosis with an associated chronic myelogenous leukaemia in a 52-year-old male patient; note one CML-typical dwarf megakaryocyte slightly left and upper to the centre of the microphotograph, one typical mastocytosis lesion and considerable eosinophilia. The diagnosis of systemic mastocytosis would have been missed without the bone marrow biopsy.
Myeloproliferative Neoplasm
Except for allogeneic haematopoietic cell transplantation (see later), which is considered potentially curative, in most myeloproliferative neoplasm (MPN) patients the treatment intention is not to achieve a cure [31]. With non-curative treatment strategies (mainly hydroxyurea) of intermediate- and high-risk patients, BM cellularity decreases, although the haematopoiesis retains its proliferative aspect and typical megakaryocytic abnormalities persist. Histological response criteria have been proposed, the cornerstones being age-adjusted BM cellularity and grade of myelofibrosis [32]. Notably, since the latter seems to be of central importance also in the treatment response estimation for JAK2-selective TKI [33, 34], standardized assessment and reporting of both reticulin fibrosis and collagen fibrosis, applying Gömöri and Masson trichrome stains (Figure 18.8) and using the established scoring criteria, is unsurpassed [35, 36].
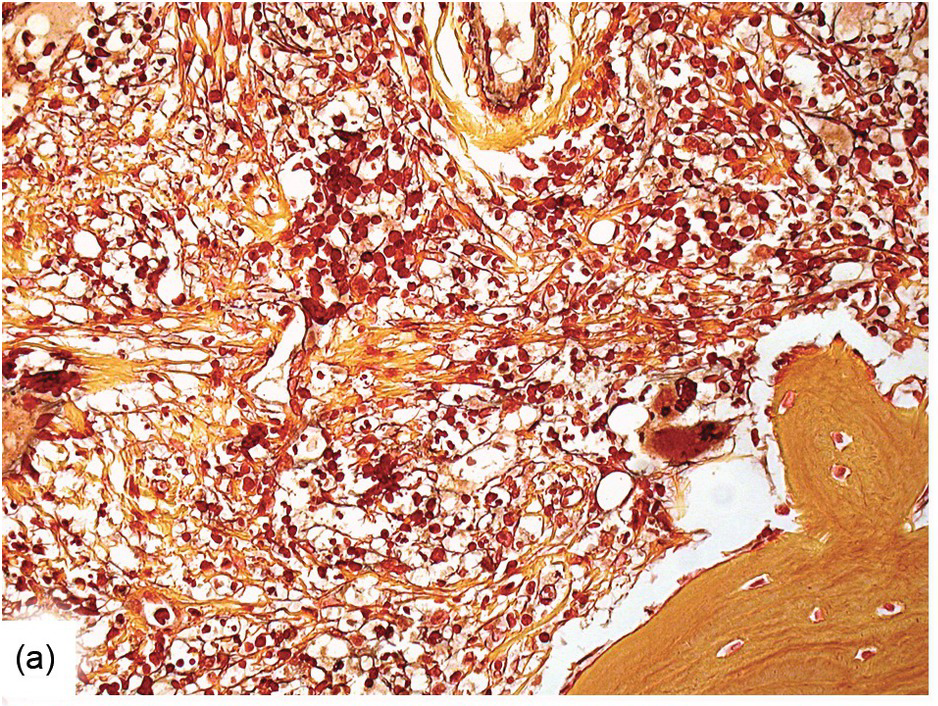
(a) Before transplantation his myelofibrosis has been scored grade 3 containing thick collagen bands and intersecting argyrophilic fibres.
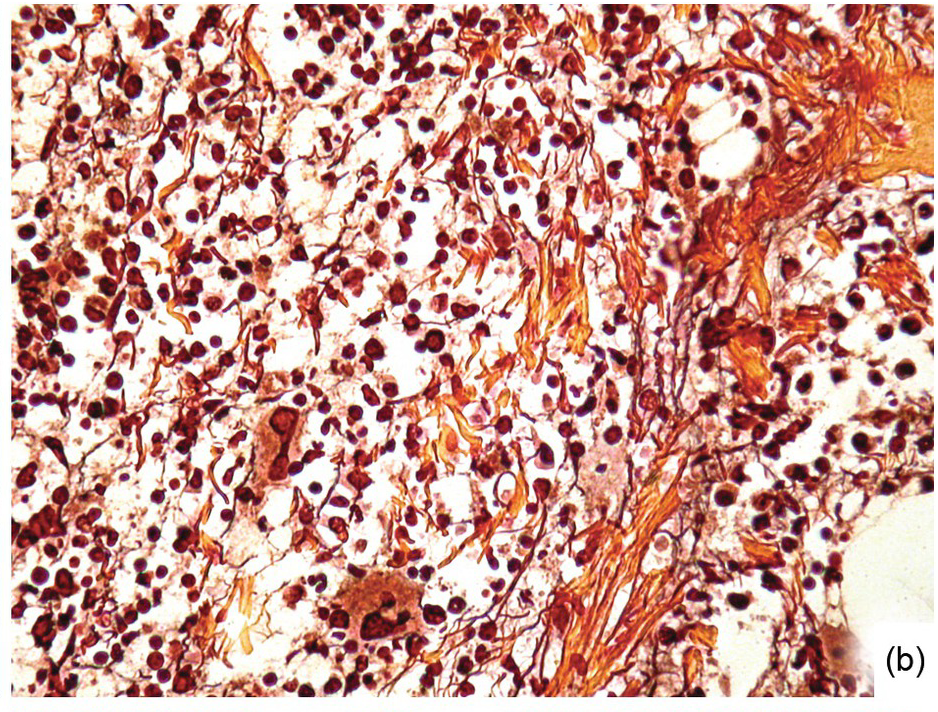
(b) Three months later his argyrophilic fibre content and intersections decreased, while collagen bands were still present.
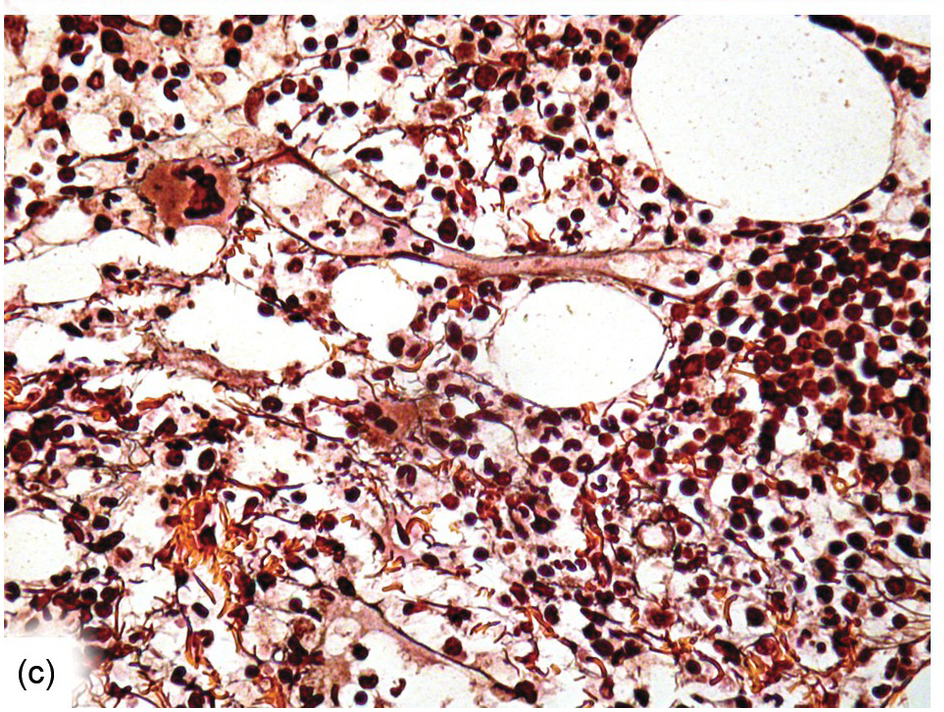
(c) The gradual decrease of both argyrophilic and collagenous fibrosis is still notable six months after transplantation.
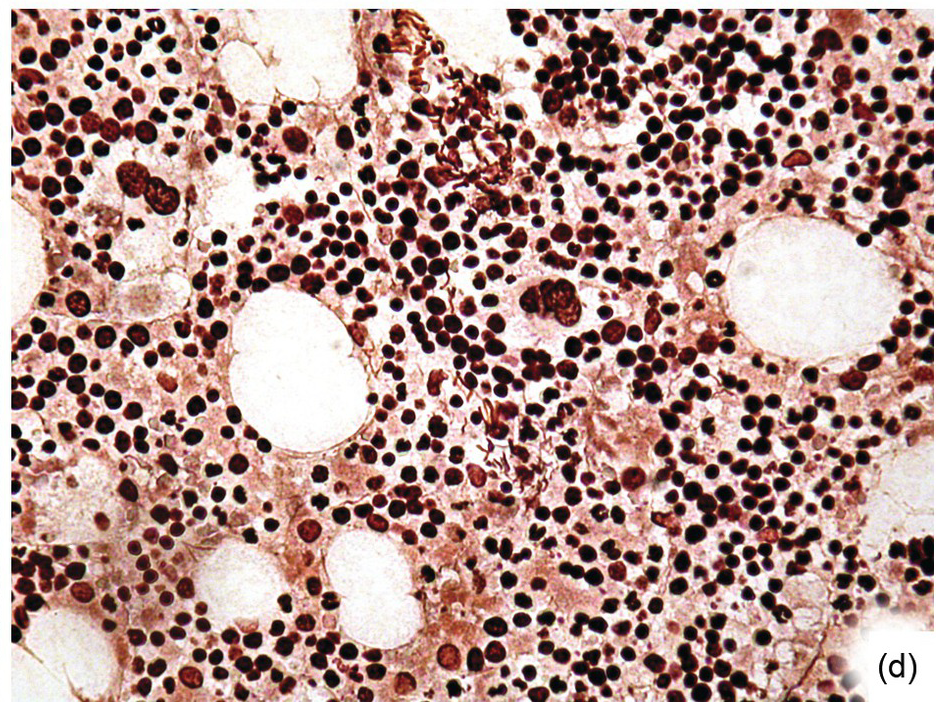
(d) One year later, the bone marrow fibre content and architecture have completely normalized; manual Gömöri staining.
Ancillary examinations based on the quantitative detection of clones bearing the relevant disease-defining mutations, i.e. JAK2 V617F, C-MPL W515L/K or CALRET type 1 or 2 mutations, by either sequencing techniques, which are feasible on paraffin-embedded BMB [37], or by IHC-based detection of CALRET mutant megakaryocytes [38] or phospho-STAT5 positive megakaryocytes may be helpful in clarifying equivocal histological remissions (Figure 18.9). Since both JAK2 and C-MPL mutant proteins activate downstream STAT5, nuclear megakaryocytic phospho-STAT5 positivity is an established marker for presence of respectively mutant clones [39, 40].
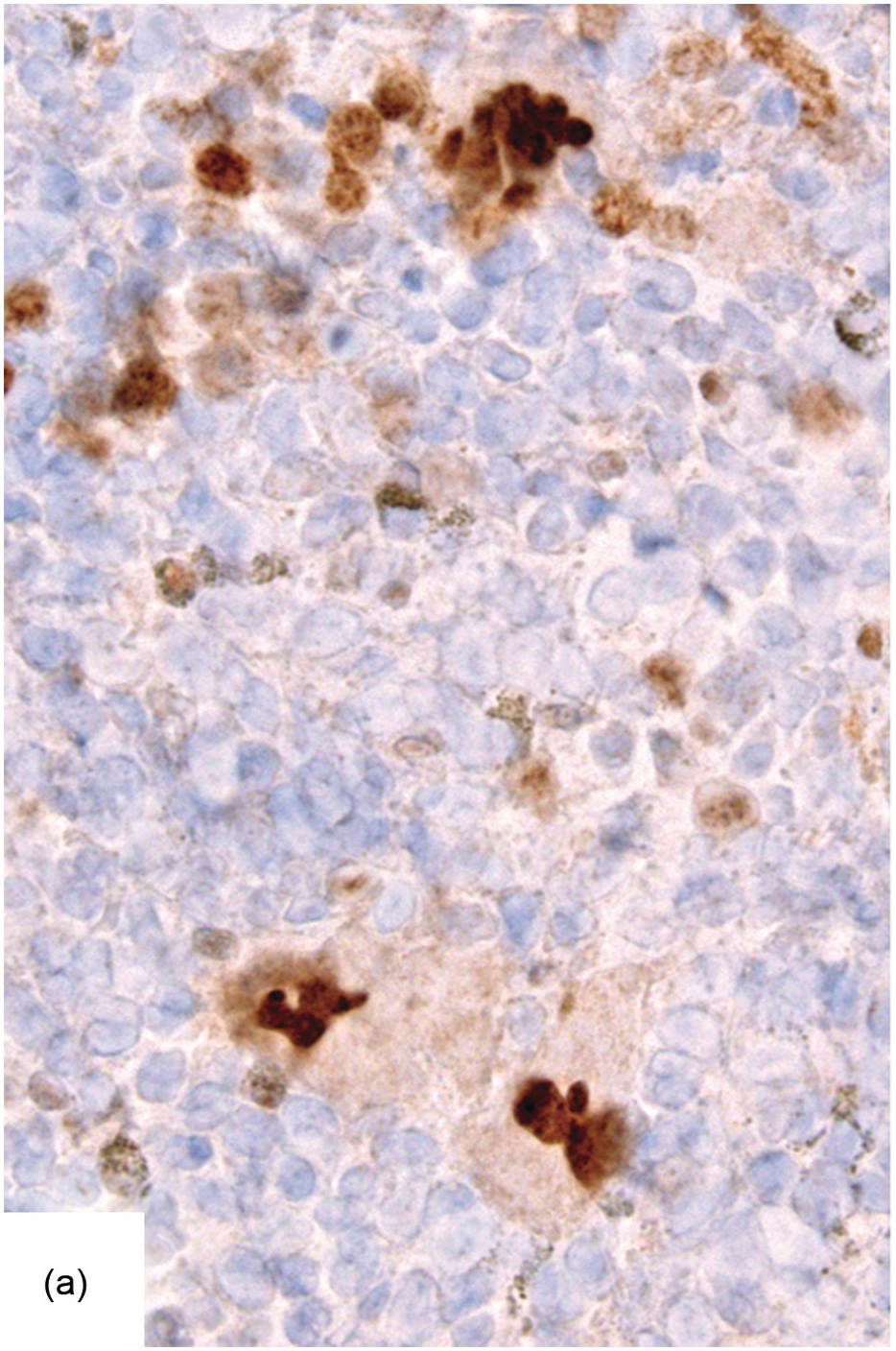
(a) Phospho-STAT5 positive megakaryocytes in a myeloproliferative neoplasm compared to their negative counterparts in a reactive condition.
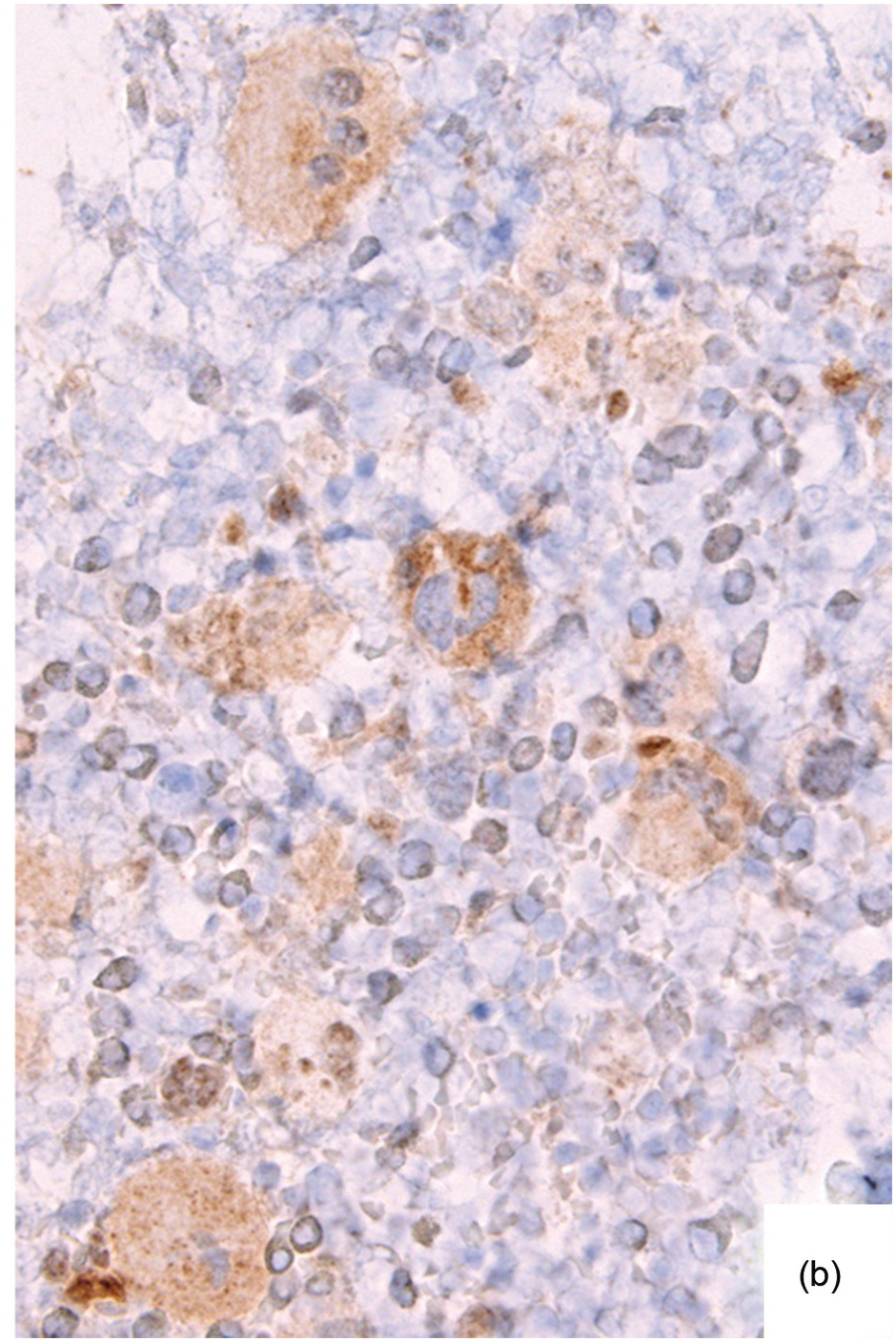
(b) JAK2 and C-MPL, and – in my personal observations – calreticulin mutant proteins activate downstream STAT5, accompanied by immunohistochemically detectable levels of phospho-STAT5 in the megakaryocytic nuclei, which turn out to uniformly and more strongly (compared to the physiologically attained erythropoiesis) stain as opposed to non-neoplastic megakaryocytes, which do not display nuclear positivity.
Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms
These categories include a variety of diseases, of which not all will progress to frank AML. Affected patients may not tolerate myeloablative therapies for various reasons, and morbidity and mortality is often due to associated cytopaenias. A major therapeutic goal in many instances will therefore be disease history alteration and alleviation of complications [41, 42]. Variable strategies, from watchful waiting, through application of growth factors, immunosuppressive/immunomodulatory drugs, demethylating agents, TKI (when applicable), up to myeloablative chemotherapy and allogeneic stem cell transplantation are pursued in myelodysplastic syndromes (MDS) and myelodysplastic/myeloproliferative neoplasms (MDS/MPN) [41, 42]. To address these problems, response criteria have been established [43, 44], with histopathological assessment playing only a minor role.
For the haematopathologist, the most important aspect is to assess absolute and relative quantitative blast changes [44] on post-treatment BMB, applying the most blast-specific markers, mainly CD34. A more challenging task is to distinguish between residual disease/residual dysplasia and reactive changes. Since this is poorly reproducible the recommendation is to accept complete disease remission regardless of dysplastic changes [43]. Megaloblastic erythropoiesis as well as dyserythropoiesis and pyknosis of megakaryocytes are non-specific and most probably related to treatment. However, presence of micromegakaryocytes (identifiable by specific staining), detection of ALIP (rigorously defined as clusters of ≥3 CD34+ blasts) or persistent strong expression of p53 in >5% of cells [45] at diagnosis and after therapy favours residual disease (Figure 18.10). Generally reversible therapy-related atypia should disappear within three months after therapy stops.
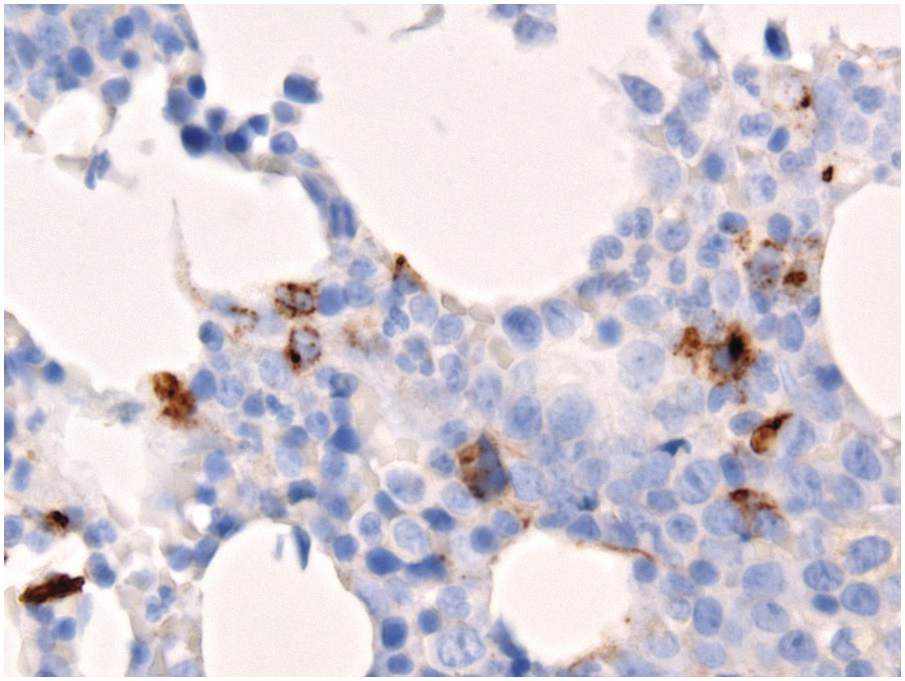
Particular attention should be paid to systematic application of p53 staining in follow-up BMB of MDS patients, particularly MDS with 5q–, who are treated with lenalidomide, since abnormal p53 protein expression (indicative of gene mutations) is linked to poor response/resistance to lenalidomide and high risk of transformation [46].
Plasma Cell Myeloma
Response criteria in plasma cell myeloma (PCM) have been established [47] and updated [48]. In this update, next-generation sequencing and next-generation flow were promoted to central importance, yet the backbone of the former criteria is still valid. Importantly, with respect to response to treatment, the highest value of BM plasma-cell infiltration assessed by any method counts. Together with the rather poor aspirability of plasma cells, this explains why histopathological BMB evaluation plays a crucial role in therapy response evaluation of PCM.
Negative immunofixation on the serum and urine combined with disappearance of any soft tissue plasmacytomas and <5% BM plasma cells define complete remission, while all the above combined with normal free light chain ratios and absence of clonal cells in BM by immunohistochemistry define stringent complete remission. Importantly the presence/absence of clonal plasma cells is based upon the immunohistochemically determined κ:λ-ratio; an abnormal ratio reflecting presence of a clone being defined in this setting as κ:λ-ratio of ≥4:1 or ≤1:2 (Figure 18.11). This is clearly different from the diagnostic criteria considering light-chain restriction for primary diagnosis of PCM with a clear-cut κ:λ-ratio of ≥15:1 or ≤1:10 [3]. Clonality of low-level plasma cell infiltrates is difficult to assess via standard immunohistochemistry for light chains and simultaneous in situ double-staining with different colour-readout is, in my view, most suitable. Expression of cyclin D1, CD56 and CD117 in the neoplastic plasma cells may also be helpful.
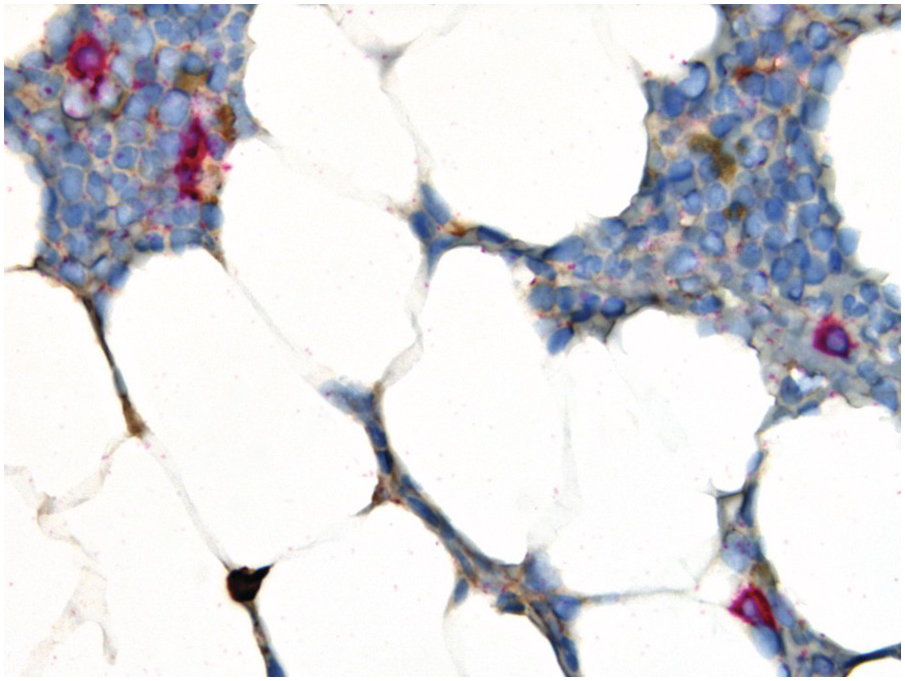
Figure 18.11 Morphological, but not stringent remission of a λ-clonal (red-counterstain) plasma cell myeloma after therapy; note that despite the paucity of plasma cells (4 to 5%) there are four visible λ+ plasma cells and one visible κ+ plasma cell, i.e. κ:λ-ratio 1:4, which is not compatible with stringency of remission.
Chronic Lymphocytic B-Cell Leukaemia
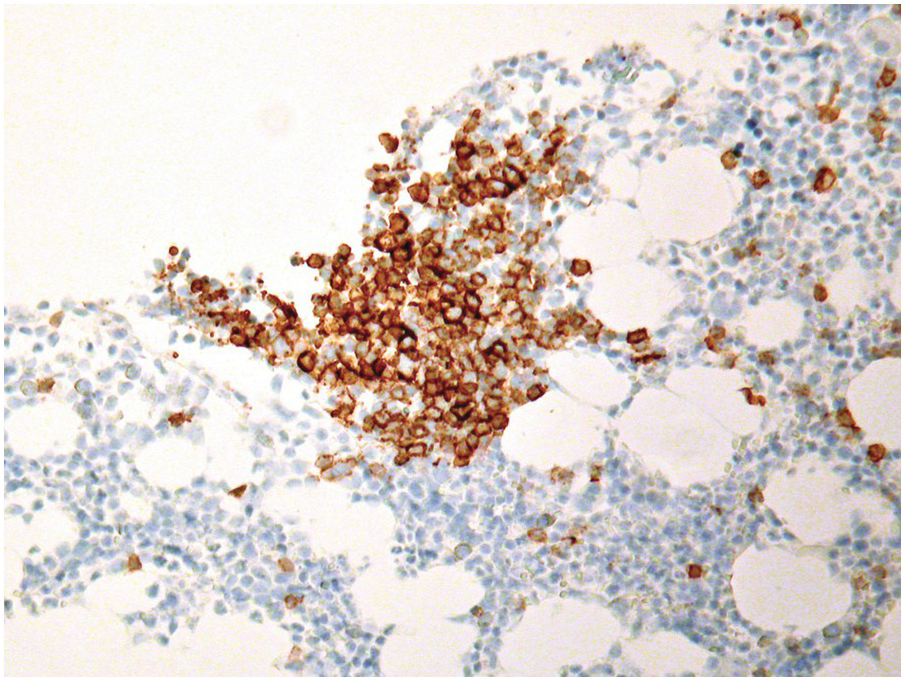
In chronic lymphocytic B-cell leukaemia (B-CLL), primary clinical and laboratory parameters are used for determination of remission [49]. Among other parameters, to assign the category of complete remission, the BM should be analyzed by FCM and/or IHC to demonstrate that it is free of (clonal) B-CLL cells. Cases with residual B-CLL cells by FCM or IHC are defined as partial remission. Since most B-CLL cases are treated with rituximab combinations, and B-CLL tends to be CD20dim positive, CD20 staining may be negative. CD5 expression might be difficult to evaluate because of tumour-infiltrating CD5-positive T-cells after therapy. A broader panel including particularly CD23, which is in my experience one of the most reliable markers in such circumstances, and CD79a and/or PAX5 must be applied (Figure 18.12). The above-mentioned response criteria are applicable also for ibrutinib-based regimens [50].
Lymphomas
Although BMB are usually performed at initial lymphoma diagnosis as a staging procedure, they can be carried out in the follow-up period in order to assign complete remission (mostly in cases that have already been involved at initial diagnosis), to detect minimal residual disease, disease progression or causes of cytopaenias, and for BM evaluation before second-line treatments such as haematopoietic stem cell transplantation. Histopathological assessment is considered very sensitive. In over 20% of cases the trephine biopsy is the only diagnostic specimen where lymphoma will be detected [3, 51, 52]. In cases with unequivocal morphology (e.g. mature B-blasts in DLBCL) or phenotype (e.g. cyclin D1 and/or SOX11 positivity in mantle cell lymphoma, CD103 and/or BRAF V600E positivity in hairy cell leukaemia, moderate to strong PD1 positivity in angioimmunoblastic T-cell lymphoma), detection of minimal residual and low-level disease can be easily achieved even if the tumour cell burden is low (Figure 18.13). However, in cases with limited involvement by lymphoma without a specific phenotype, such as marginal zone lymphoma for example, detection of minimal residual disease can be very difficult and can be improved by applying modern molecular methods [53].
Effects of Drugs Targeting Haematopoietic Signalling and Specific Molecules on/in Haematolymphoid Cells
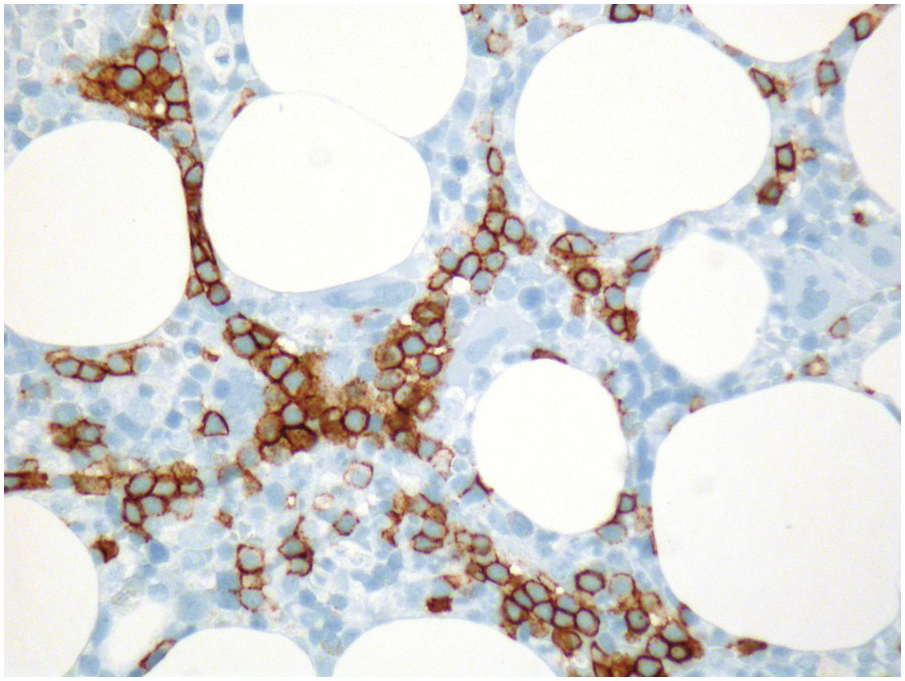
Growth factor signalling is central to BM function. Thus targeting this signalling pathway inevitably leads to predictable morphologic BM changes. Granulocyte- and, rarely, granulocyte-macrophage colony-stimulating factors (G-/GM-CSF), given for the treatment of congenital and acquired neutropaenias, for mobilization of peripheral haematopoietic progenitor cells for stem cell transplantation and to foster recovery after myelotoxic chemotherapy, induce an overall increase in cellularity and, in particular, a left shift of myelopoiesis with increased myeloblasts and monoblasts, resembling AML or CMML but being properly localized along the trabeculae [54, 55] (Figure 18.14). Rarely, increased haemophagocytosis [56] and BM necrosis [57] have been reported. In addition, tumours with paraneoplastic production of G-CSF have been observed to cause MPN-like states [58]. A diagnostic challenge in post-treatment BM samples of AML patients who have received G-CSF is to distinguish reactive myeloid hyperplasia from residual disease. For obvious reasons this cannot be achieved without explicit communication of prior growth factor therapy by the clinicians to the haematopathologist. The microarchitectural localization of blasts by IHC might be helpful since groups or sheets of blasts with non-peritrabecular distribution (ALIP) are indicative of persistent leukaemia.
Figure 18.14 Myelopoietic hyperplasia and left shift after administration of granulocyte colony-stimulating factor; note the expansion of the peritrabecular myeloid compartment and the considerable amount of immature myelocytes protruding to the centre of the marrow spaces.
Regarding possible side effects of long-term G-CSF administration, approximately 20% of severe congenital neutropaenia (SCN) patients will develop AML after long treatment with G-CSF [59]. Sequential myeloid colony assays performed at different time points during development from SCN to secondary AML, revealed early disease driving mutations in the CSF3R gene, the clone being given additive survival advantage by exogenous G-CSF rather than G-CSF being directly leukemogenic [60]. There is no evidence that G-CSF can cause AML in healthy donors [61].
Thrombopoietin and thrombopoietin receptor agonists given to patients suffering from idiopathic thrombocytopaenic purpura or, rarely, in aplastic anaemia, are able to induce transient morphologic changes resembling MPN (Figure 18.15), including myelofibrosis [62, 63]. As expected, paraneoplastic production of thrombopoietin will also lead to thrombocytosis [64].
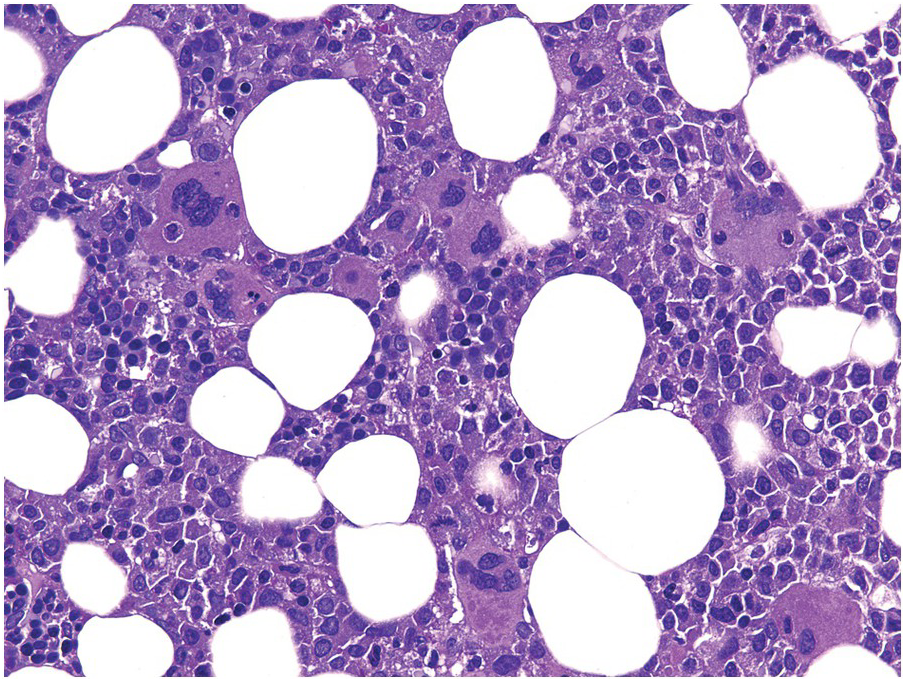
Erythropoietin therapy for chronic anaemia, e.g. in chronic renal failure, and in appropriate MDS cases, can be accompanied by erythroid hyperplasia with non-specific megakaryocytic- and erythroid changes [65]. Endogenous erythropoietin production in a paraneoplastic state called telangiectasias, elevated erythropoietin level and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting (TEMPI)-syndrome [66], will produce similar changes.
JAK-inhibitors [67] applied for the treatment of MPN, chronic inflammatory and autoimmune diseases, as well as graft versus host disease (GvHD, see later), inhibit downstream signalling of all above-mentioned haematopoietic growth factors and cytokines, and therefore can often cause anaemia and thrombocytopaenia [68]. While in MPN, a clear-cut histomorphologic correlation will rarely be perceptible, in non-neoplastic conditions, a hypocellularity with erythroid and megakaryocytic hypoplasia can be observed (Figure 18.16).
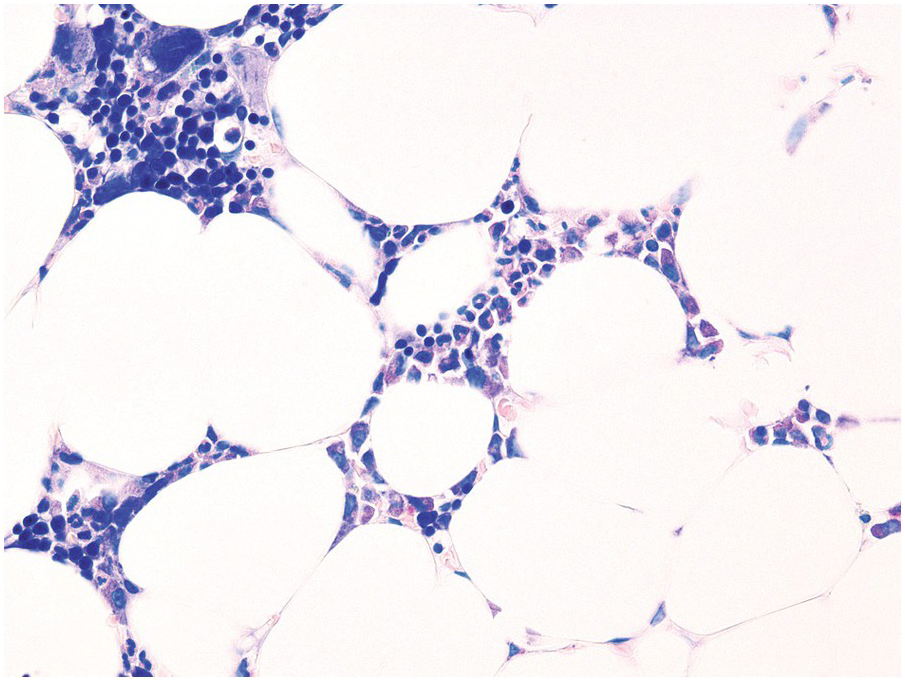
Figure 18.16 Bone marrow hypocellularity with erythroid and megakaryocytic hypoplasia in a patient, who has been treated with the JAK1-inhibitor itacitinib for graft versus host disease (Giemsa).
Interferon-α, which is approved, although rarely used, for the treatment of hairy cell leukaemia, B-cell lymphomas, melanoma, CML and MPN, renal cell carcinoma, neuroendocrine tumours and chronic viral hepatitis, to name a few [69], can induce sarcoid-like BM granulomas in addition to occasional profound sustained myelosuppression (Figure 18.17) [70].
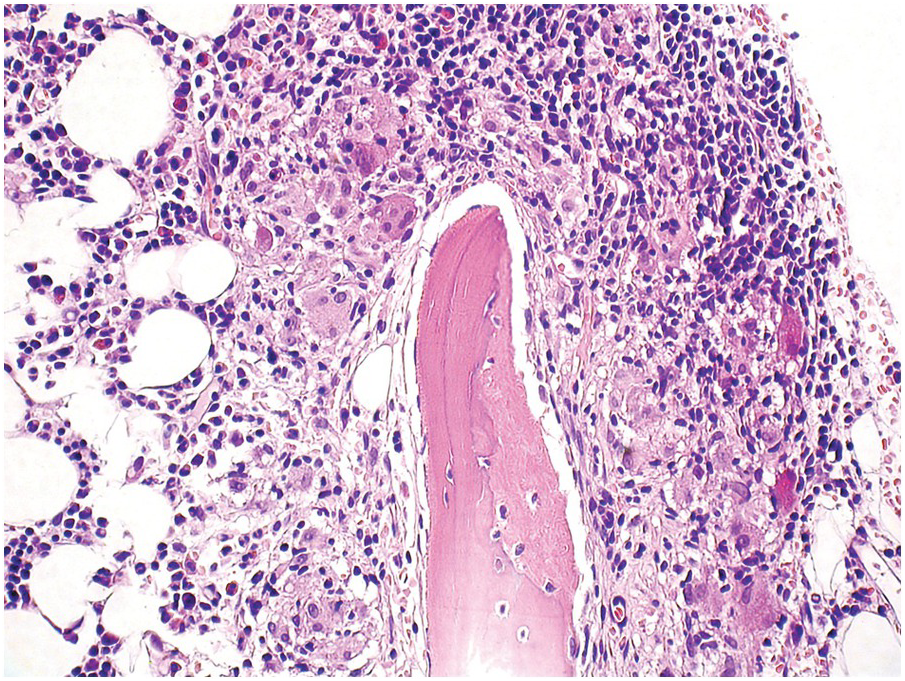
Figure 18.17 Bone marrow granuloma formation in a low-grade lymphoma patient, who has been treated with interferon-α.
Immunomodulatory agents, such as lenalidomide and derivatives, are widely used in the treatment of PCM [71], MDS subtypes [72] and, recently, in some lymphomas [73]. Application of this type of drug is initially accompanied by remarkable degenerative changes in the neoplastic plasma cells with cytoplasmic vacuolization and fragmentation, increased apoptotic bodies and, finally, in therapy-sensitive cases, complete morphologic remission. In therapy-sensitive MDS, complete resolution of dysplasia, especially disappearance of typical 5q– megakaryocytes, can be expected. As discussed above, the importance of p53 staining in newly diagnosed MDS patients suitable for lenalidomide therapy and in follow-up biopsies cannot be overemphasized [46].
Antibodies targeting CD20, such as rituximab, are used either as single agents or, more commonly, in combination-therapy regimens, in almost all B-cell lymphomas and for a few inflammatory/autoimmune disorders [74]. Treatment is very effective and usually eradicates B-cells/B-cell lymphomas in the BM, inducing or leaving nodular lymphoid infiltrates containing solely T-cells (Figure 18.18a), of which one should be aware so as not to overdiagnose residual lymphoma [75]. During anti-CD20 targeting, mostly transient CD20-negative tumours/tumour cell subclones may evolve [76, 77]. This has an immediate practical diagnostic impact, requiring application of alternative B-cell markers such as CD19, CD79a or PAX5 to identify residual disease. Known side effects of rituximab are mostly self-limiting, late onset neutropaenia [78, 79] that is reflected by a myeloid maturation arrest in the BM (Figure 18.18b), thrombocytopaenia and, rarely, disseminated intravascular coagulation [80].
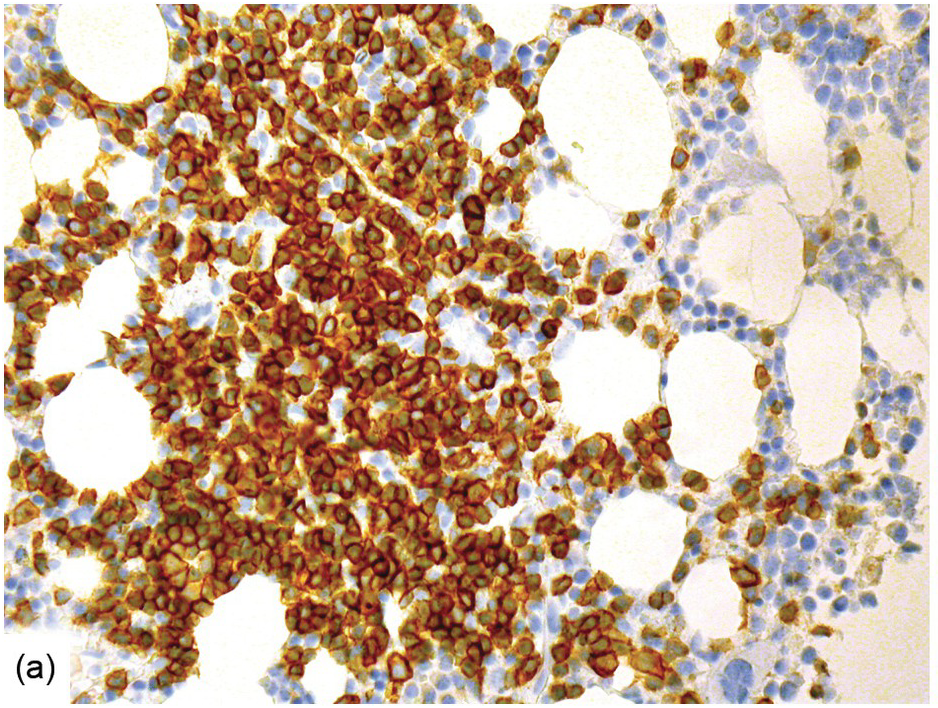
(a) Nodular lymphoid infiltrate containing solely T-cells as highlighted by CD3 in a follicular lymphoma patient, who has been given rituximab.
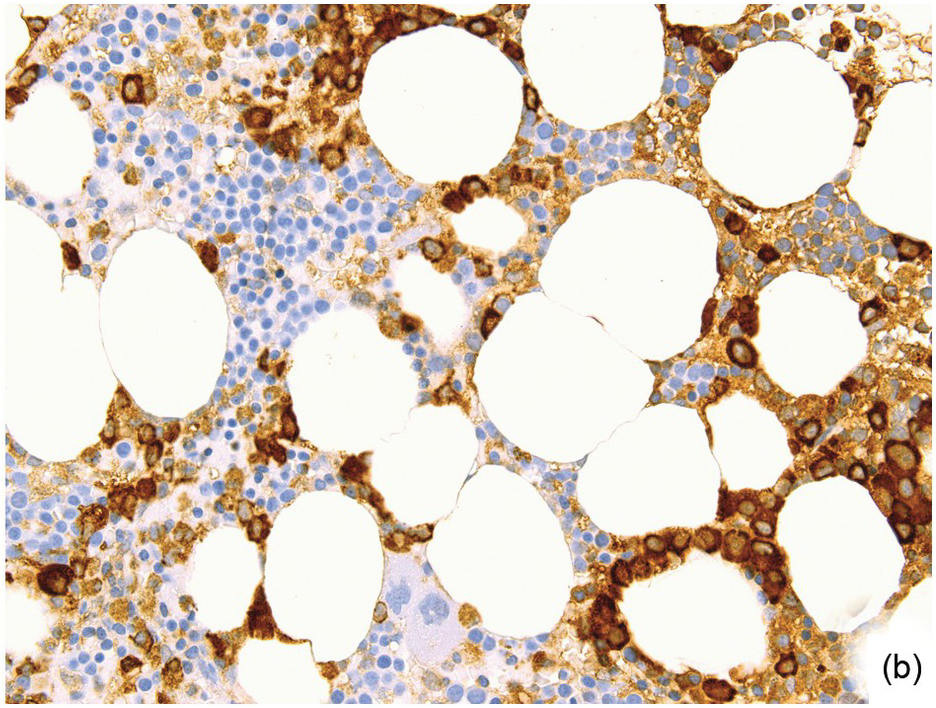
(b) Regenerating myelopoiesis with ‘myeloid maturation arrest’ after rituximab.
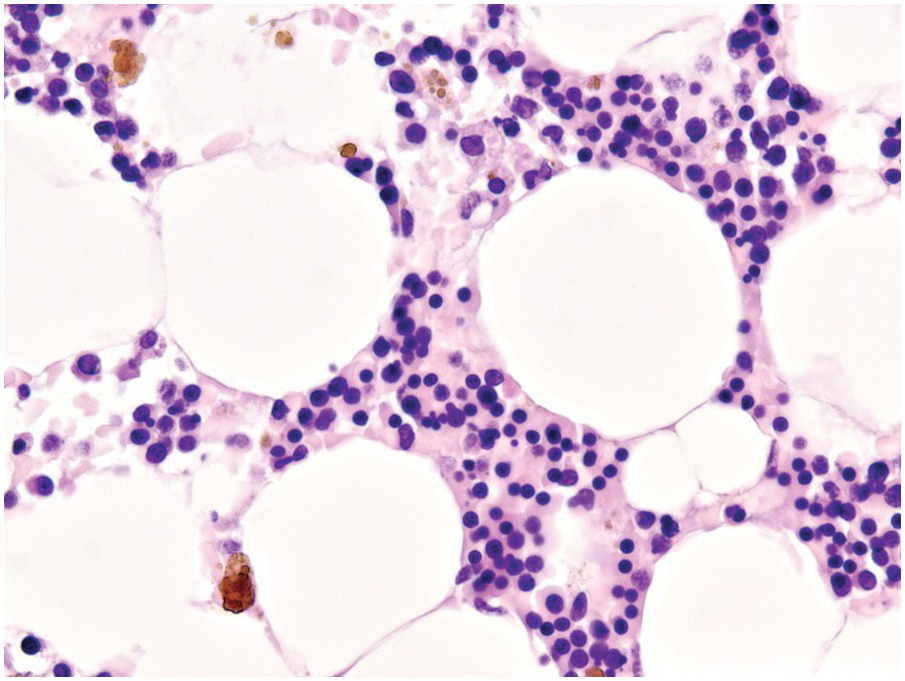
The drug-conjugated monoclonal antibody targeting CD30, brentuximab vedotin, is approved for the second-line treatment of classical Hodgkin lymphoma and anaplastic large cell lymphoma, and may soon become part of some primary treatment regimens for these diseases [81]. A very common (20% of patients) side effect of brentuximab is neutropaenia that is multifactorial and synergistic to co-administered chemotherapy [82]. There are no studies on the BM morphology in such cases. In an autopsy case, I was able to observe hypocellularity, decreased and left-shifted myelopoiesis as well as severe dyserythropoiesis (Figure 18.19).
The monoclonal antibody targeting CD38, daratumumab, is efficient in relapsed/refractory myeloma, and emerging data from clinical trials will likely result in its approval as first-line treatment of PCM in combination with other drugs [83]. Following anti-CD38 targeting CD38-negative PCM relapses may evolve [84] for the diagnosis of which alternative antibodies such as those against CD138 and/or MUM1p should be considered.
The monoclonal antibody binding to CD52, alemtuzumab, is approved for the first-line treatment of T-prolymphocytic leukaemia, as well as second-line treatment for B-CLL, in some transplant settings and for selected cases of multiple sclerosis [85]. Since CD52 is present on normal and malignant B-, T- and NK cells, on macrophages, monocytes and granulocytes, alemtuzumab leads to antibody-dependent cell lysis of all of these. Lymphopaenia with increased risk of opportunistic infections, pancytopaenia and immune thrombocytopaenia are expected side effects that can be reflected in BM changes.
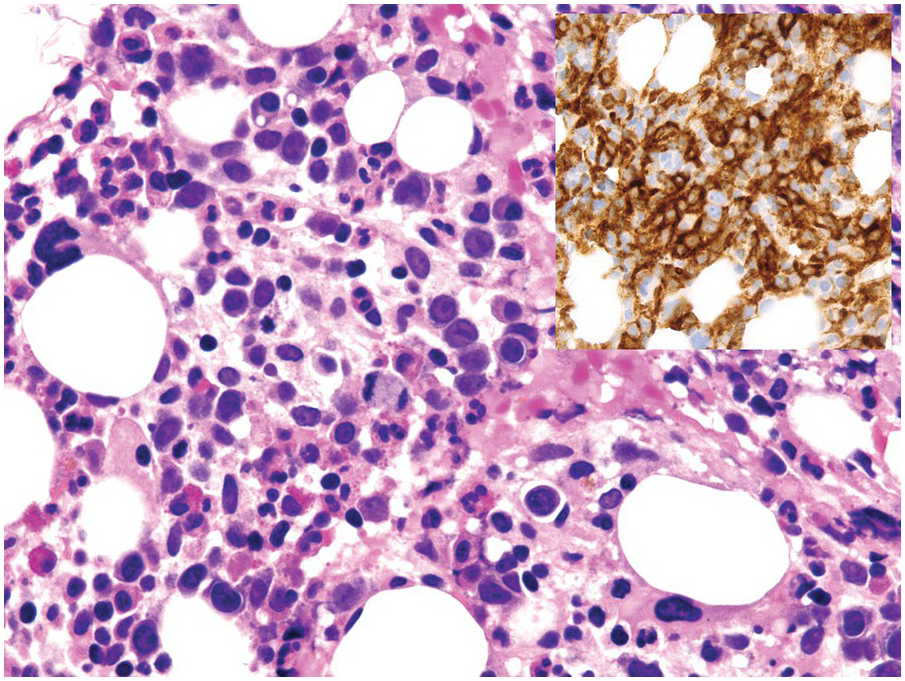
Patients with multiple sclerosis on treatment with the integrin α4β1-targeting monoclonal antibody, natalizumab, can suffer from severe, but reversible, hyporegenerative anaemia [86, 87]. Natalizumab [88] induces erythroid maturation arrest in the BM with the consequent clinical picture of severe non-regenerative, non-haemolytic anaemia, and an increase of monocytic precursors reminiscent of CMML (Figure 18.20), which is expected considering the mechanism of its action in hindering leukocyte migration across barriers. Importantly, a monoclonal antibody targeting the α4β7 (intestinal) integrin, vedolizumab, is not linked to BM abnormalities.
Despite the common observation of peripheral neutropaenia and lymphocytosis in up to a quarter of ibrutinib-treated patients [89], there are no reports on any specific morphologic BM changes.
Azathioprine, Mercaptopurine, Thioguanine, Allopurinol and Thiopurine S-Methyltransferase Deficiency
All the above-mentioned drugs, given for a variety of diseases, ranging from ALL, through autoimmune/idiopathic inflammatory diseases to gout, have in common either being metabolized by or inhibiting, in the cases of allopurinol, the enzyme thiopurine-S-methyltransferase (TPMT) [90]. The effects of these drugs are achieved through incorporation of their active metabolites thiodeoxyguanosine triphosphate into DNA and thioguanosine triphosphate into RNA, by inhibition of de novo purine synthesis by methylmercaptopurine nucleotides, and by inhibition of Rac1 to induce apoptosis in activated T-cells [91]. From the BM perspective, this is reflected in various erythroid abnormalities including dyserythropoiesis and megaloblastic changes, dysmegakaryopoiesis (small pyknotic forms with hypo- or alobated nuclei) and nuclear segmentation abnormalities of myelopoiesis [3, 92]. The lifespan of these metabolites is determined by the proper function of TPMT. It is reported that approximately 10% of the population has reduced TPMT activity and approximately 0.5% have a true deficiency of TPMT [93]. The latter are at risk of potentially life-threatening BM toxicity with conventional drug dosages, which can be aggravated by concomitant application of allopurinol. This is accompanied by pancytopaenia, severe hypoplasia, increased intramedullary apoptosis and considerable dysplastic changes in the erythroid and megakaryocytic lineages (Figure 18.21).
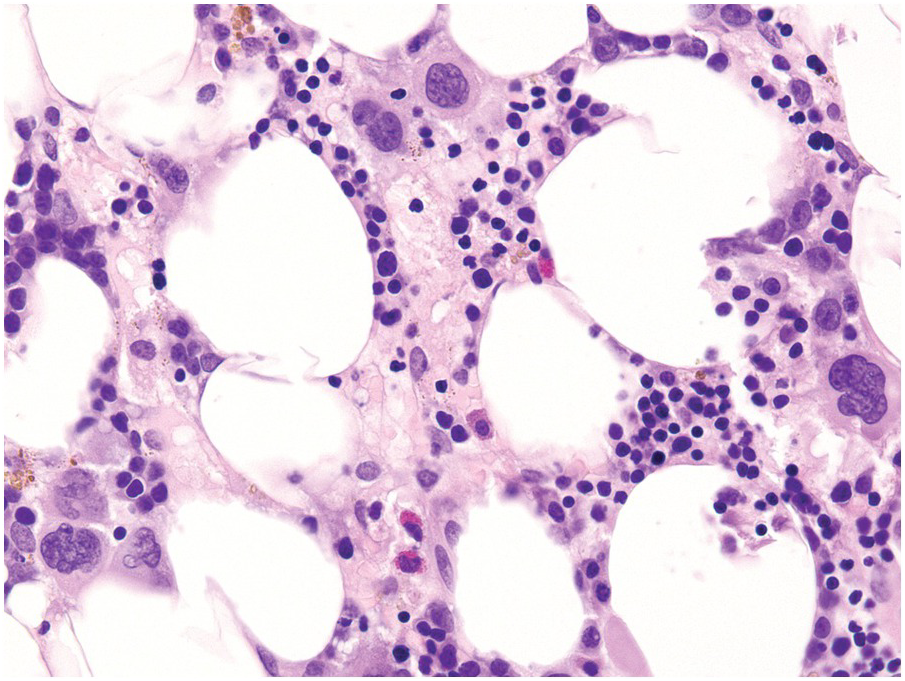
Figure 18.21 Considerable dysplastic changes in the erythroid and megakaryocytic lineages in a patient exposed to azathioprine; note the increase of small atypical megakaryocytes.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


