Abstract
Plasma cell neoplasms are derived from mature, IG heavy chain class-switched terminally differentiated B-cells, which usually secrete a monoclonal immunoglobulin or M-protein and consist of a homogeneous population of neoplastic plasma cells [1]. Plasma cell myeloma (PCM) is a common malignancy, manifesting itself by bone marrow (BM) infiltration and bone destruction, and therefore representing an entity frequently encountered in BM biopsies(BMB). Its precursor lesion, non-IgM monoclonal gammopathy of unknown significance (MGUS) is a frequent finding in elderly individuals and shows a low, but definite risk for progression to PCM. In this chapter, we will discuss the diagnosis and differential diagnosis of PCM, MGUS and clonal plasma cell disorders with associated paraneoplastic syndromes, including POEMS syndrome and TEMPI syndrome. Amyloidosis is discussed in Chapter 3, and other mature B-cell neoplasms with a clonal plasma cell component in Chapter 15.
Introduction
Plasma cell neoplasms are derived from mature, IG heavy chain class-switched terminally differentiated B-cells, which usually secrete a monoclonal immunoglobulin or M-protein and consist of a homogeneous population of neoplastic plasma cells [1]. Plasma cell myeloma (PCM) is a common malignancy, manifesting itself by bone marrow (BM) infiltration and bone destruction, and therefore representing an entity frequently encountered in BM biopsies(BMB). Its precursor lesion, non-IgM monoclonal gammopathy of unknown significance (MGUS) is a frequent finding in elderly individuals and shows a low, but definite risk for progression to PCM. In this chapter, we will discuss the diagnosis and differential diagnosis of PCM, MGUS and clonal plasma cell disorders with associated paraneoplastic syndromes, including POEMS syndrome and TEMPI syndrome. Amyloidosis is discussed in Chapter 3, and other mature B-cell neoplasms with a clonal plasma cell component in Chapter 15.
Non-IgM Monoclonal Gammopathy of Unknown Significance
Definition and Epidemiology
Non-IgM monoclonal gammopathy of unknown significance (MGUS) is a very common premalignant precursor lesion of plasma cell neoplasms [2–7]. It is defined as the presence of complete or incomplete (isolated kappa or lambda light chains) clonal immunoglobulin in the serum and/or urine. The paraprotein is usually of IgG (60%) or IgA (15%) type, rarely IgD, IgE (1% each) or biclonal (3%). MGUS with IgM paraprotein is usually a precursor lesion of lymphoplasmacytic lymphoma/Waldenström’s disease and frequently carries the pathognomonic MYD88 L265P mutation [1, 8]. By definition, the serum M protein concentration is less than 30 g/L, clonal plasma cells are <10% in the BM and end-organ damage including the so-called CRAB criteria (hyperCalcaemia, Renal insufficiency, Anaemia and Bone lesions) and AL amyloidosis are absent, and there is no evidence of a solitary plasmacytoma. Light chain MGUS accounts for 20% of cases and is defined by an abnormal free light chain ratio and urinary light chain excretion <0.5 g/24 hours [2, 4].
Monoclonal gammopathy of unknown significance occurs in 3 to 4% of individuals above 50 years of age and it is usually detected during routine serum electrophoresis [6]. The rate for progression to PCM, or infrequently to AL amyloidosis or small B-cell lymphoma, is approximately 1% per year. Although the risk for progression is very difficult to assess for an individual patient, plasma cell percentage in the BM, paraprotein level, free light chain ratio and the type of paraprotein (IgG versus IgA), as well as phenotypic abnormalities identified by flow cytometry, may help to identify patients at risk who may need closer follow-up [4, 9].
Clinical Features
By definition, individuals with non-IgM MGUS are asymptomatic and lack the typical laboratory and radiological features of PCM. It is detected during screening or laboratory work-up for another disorder. About 30 to 40% of patients have a reduction of the other immunoglobulins in the serum.
Morphology
Individuals with MGUS usually show no or a discrete increase of usually mature plasma cells in the BM, which cannot be differentiated from reactive plasma cells without additional immunohistochemical or flowcytometric studies [1, 2]. Plasma cells are distributed singly in the haematopoietic marrow or along vascular structures. Rarely plasma cells may show cytologic atypia, which would raise the suspicion of PCM, even in the absence of >10% plasma cells. Clusters of plasma cells without relation to blood vessels or in the fatty marrow should also raise suspicion for the presence of PCM.
Immunophenotype
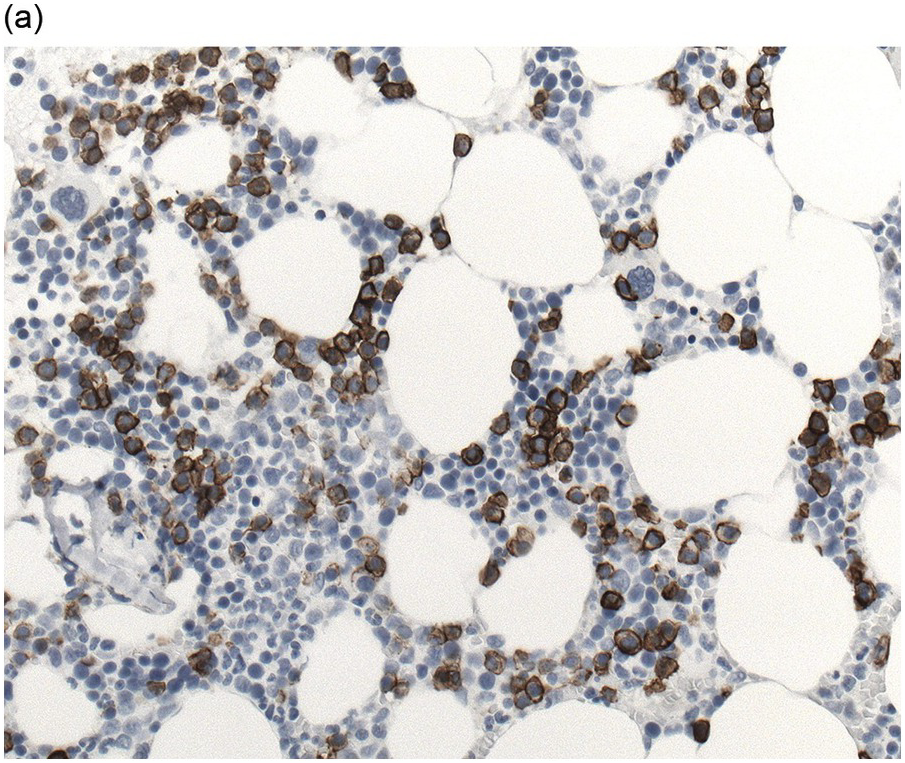
Like normal mature plasma cells, MGUS shows expression of plasma cell markers, including CD138, CD38 and MUM1, which can be used for better enumeration [10]. The kappa/lambda expression ratio can be normal or skewed towards one of the immunoglobulin light chains, depending on the amount of clonal plasma cells. Aberrant CD56 expression may be identified in a minority of MGUS, but is usually weak and only present in a minority of plasma cells [11]. Cases of MGUS with the t(11;14) translocation show strong nuclear expression of cyclin D1 in all plasma cells, a feature that can be used to confirm a clonal plasma cell population. Flow cytometry may identify abnormal immunophenotypic features of plasma cells, such as lack of CD19 [9].
Genetic Features
The genetic profile of MGUS is very similar to PCM, with almost 50% of cases showing translocations involving the immunoglobulin heavy chain gene and about 40% exhibiting a hyperdiploid karyotype [12, 13]. Of note, although one can assume that advanced PCM has a higher frequency of genetic alterations, genetic aberrations per se are not able to distinguish between MGUS and PCM and in general are not useful for prognostic purposes [14, 15]. For details on cytogenetics and molecular genetics see below.
Plasma Cell Myeloma
Synonyms
Multiple myeloma, medullary plasmacytoma, Kahler disease.
Definition and Epidemiology
Plasma cell myeloma is a BM-based, multifocal malignancy of plasma cells, usually associated with paraproteinaemia and end-organ damage as described below; cases without CRAB criteria, but surpassing the limits for MGUS as described above are designated smouldering (asymptomatic) PCM [4, 16]. The differentiation between MGUS, smouldering PCM and manifest PCM is based on a combination of clinical, laboratory and morphological features and therefore requires integration of all findings [1, 17, 18]. The diagnostic criteria for MGUS, smouldering PCM and PCM are summarized in Table 16.1.
Table 16.1 Diagnostic criteria for non-IgM monoclonal gammopathy of undetermined significance and plasma cell myeloma.
Non-IgM MGUS
Monoclonal serum protein <30 g/L or abnormal free light chain ratio (<0.26 or >1.65) and urinary M protein <500 mg/24 hours
Clonal bone marrow plasma cells <10%
No end-organ damage (hyperCalcaemia, Renal insufficiency, Anaemia or Bone lesions = lack of CRAB criteria)
Asymptomatic (smouldering) myeloma (both criteria must be met)
Monoclonal serum protein >30 g/L or urinary M protein >500 mg/24 hours and/or 10–60% clonal bone marrow plasma cells
No end-organ damage or amyloidosis
Plasma cell myeloma
>10% clonal plasma cells or biopsy-proven plasmacytoma and >1 myeloma-defining event (>1 CRAB criteria fulfilled)
>1 biomarker of malignancy
> 60% clonal bone marrow plasma cells
serum free light chain ratio >100
>1 focal lesion on MRI
PCM accounts for about 1% of all malignancies and approximately 10 to 15% of haematopoietic neoplasms [19]. It is slightly more common in males with a male to female ratio of 1.4:1 and almost twice as frequent in Black as in Caucasian populations [1, 19]. PCM is a disease of adults, and 90% of cases occur above the age of 50. It is rare below the age of 30 and only exceptionally observed in children [20].
Clinical Features, Prognostic Factors and Staging Systems
Symptomatic PCM is characterized by the presence of end-organ damage and appropriate clinical symptoms in patients with a monoclonal plasma cell population in the BM and M protein in serum or urine [1, 17, 18]. For the pathogenesis of clinical symptoms and complications, four major factors are responsible, including (1) bone destruction through activation of osteoclasts and osteoblast suppression, (2) peripheral cytopaenias through replacement of haematopoietic marrow by neoplastic plasma cells and lack of erythropoietin through kidney damage, (3) immunoglobulin deficiency through suppression of normal polyclonal plasma cells and (4) kidney damage, hyperviscosity symptoms and rarely AL amyloidosis through production of monoclonal immunoglobulins.
The most prevalent symptom is bone pain due to multiple osteolytic lesions, mostly affecting the axial skeleton and the large long bones, frequently accompanied by general weakness, pallor, haemorrhagic diathesis and an increased incidence of infection. More than 70% of patients show skeletal damage, including diffuse osteoporosis and osteolytic lesions, which can result in spontaneous fractures and paralysis. MRT and PET/CT identify bone lesions in an even higher percentage of patients [21]. Renal failure, which may be a presenting symptom, is due to tubular damage by secreted light chains, especially the lambda isotype [22]. Almost all patients show a monoclonal paraprotein in serum or urine, with IgG in >50%, IgA in 20%, light chains only (Bence Jones protein) in 15 to 20%, with rare cases showing IgD, IgE or IgM or biclonal paraproteins [1, 16, 17, 22]. Approximately 1% of cases are non-secretory. Nevertheless, in most of these cases cytoplasmic immunoglobulin can be demonstrated, pointing to defects in Ig secretion [2]. In a minority of cases, molecular alterations in immunoglobulin genes result in lack of Ig translation [23].
Infiltration of extramedullary sites is commonly a feature of advanced disease and needs to be distinguished from primary extramedullary plasmacytoma, a localized, usually indolent plasma cell neoplasm frequently arising in the head and neck region [1, 24].
Over the years, a variety of staging systems have been developed for patients with manifest PCM [25, 26]. Currently, the International Staging System (ISS) [26], complemented by cytogenetic risk factors [27], provides a powerful predictor for prognosis.
Morphology
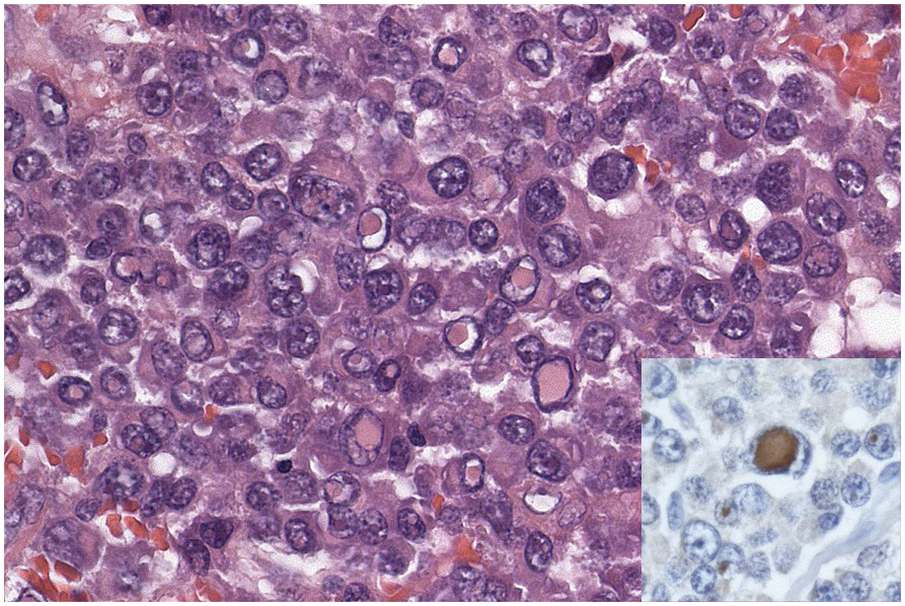
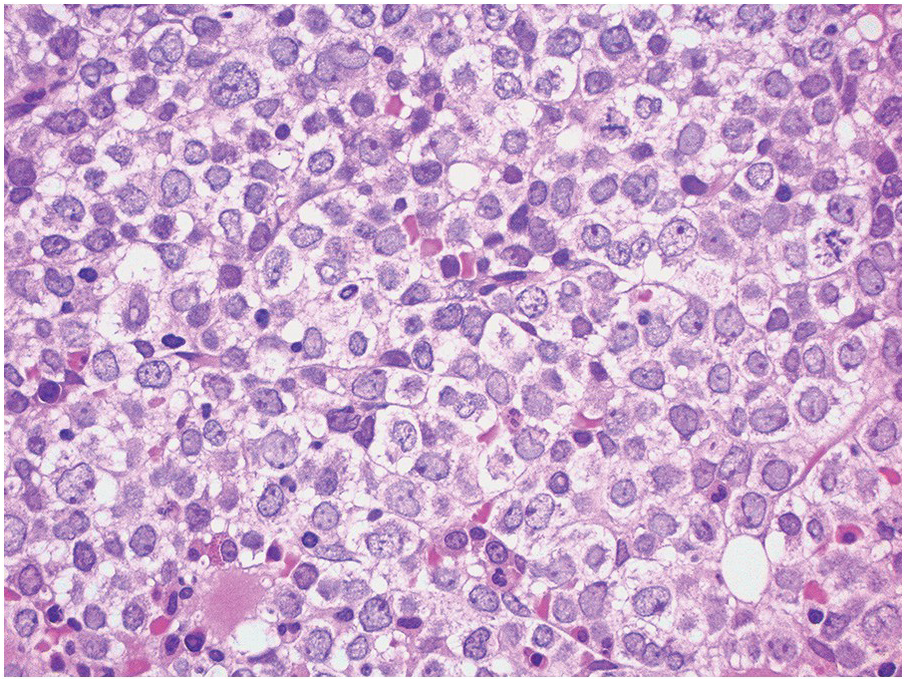
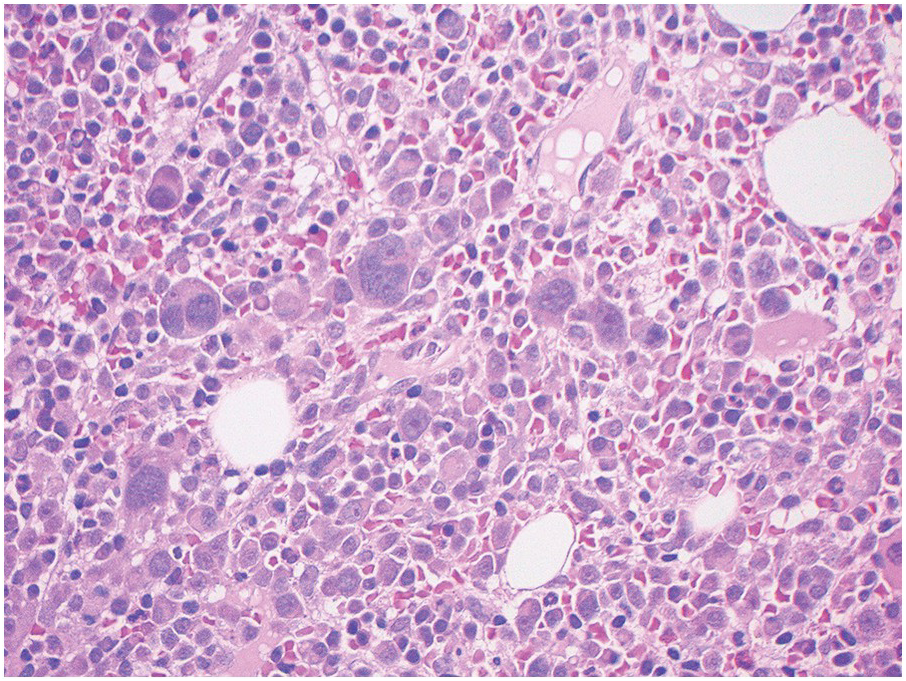
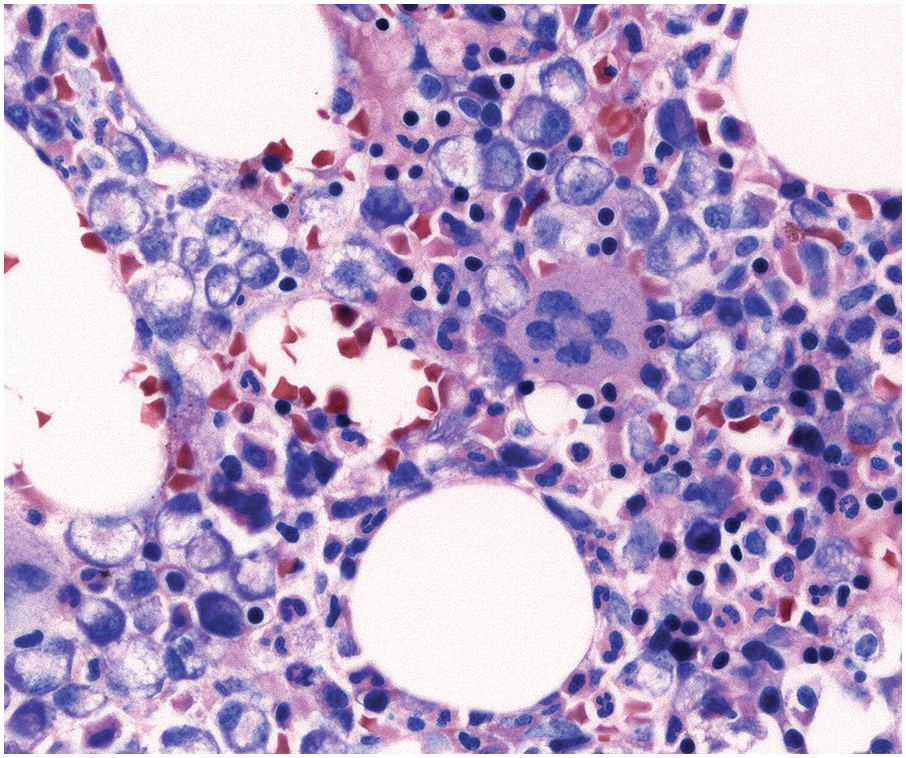
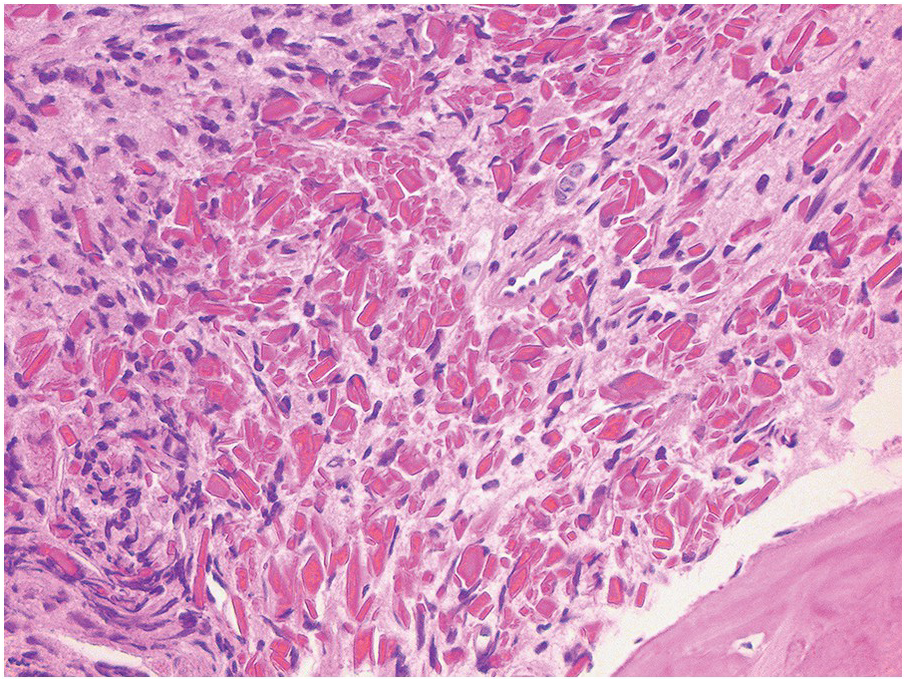
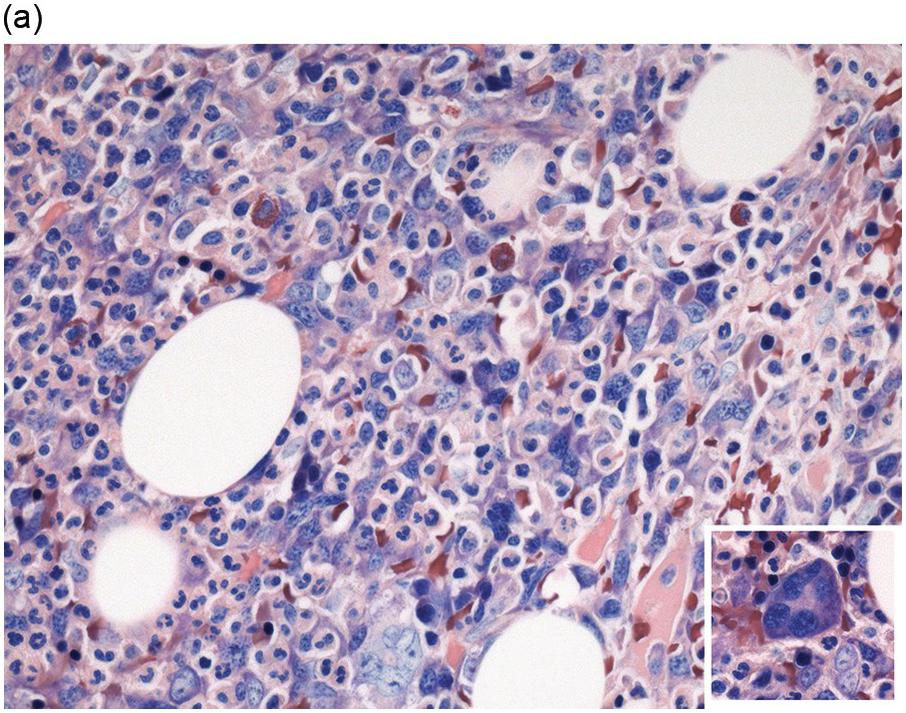
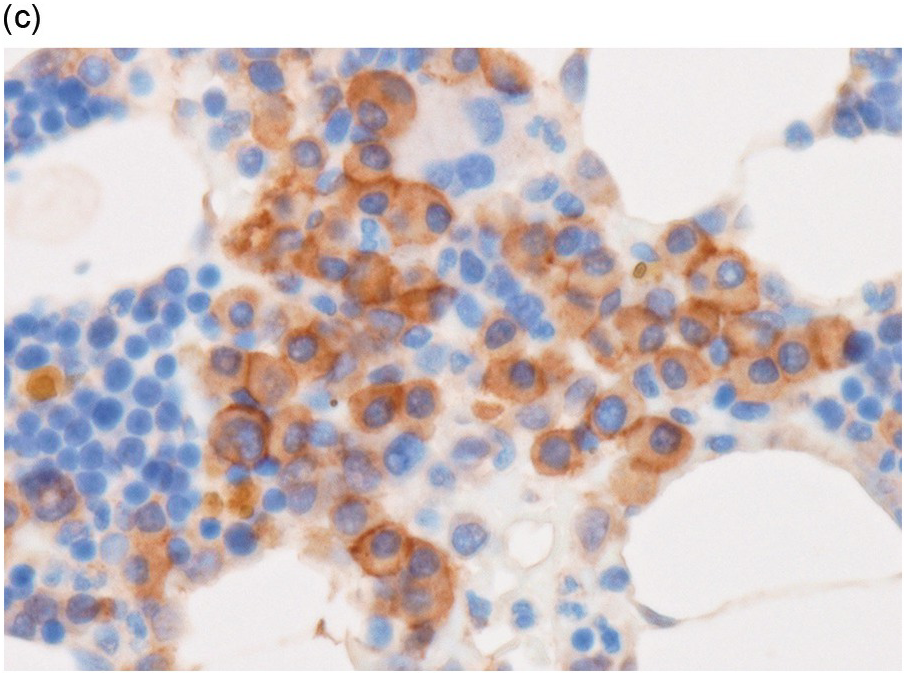
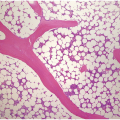
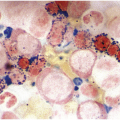
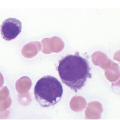
In the BM aspirate, the number of plasma cells in PCM is extremely variable from case to case. The cytomorphology of the neoplastic cells shows a broad spectrum, ranging from mature plasma cells without cytologic atypia to plasmablastic and anaplastic cells, which sometimes cannot be differentiated morphologically from aggressive non-Hodgkin lymphomas, other haematopoietic neoplasms or even metastatic carcinoma. Mature plasma cells show a typical clock-face nucleus with eccentric location and basophilic cytoplasm with a lighter stained perinuclear hof (Figure 16.1). The lymphoplasmacytic or small-cell variant of PCM shows small cells with a narrow rim of basophilic cytoplasm with a small hof, which may be difficult to distinguish from mature lymphocytes (Figure 16.2a–d); this morphology is frequently associated with the t(11;14) translocation [28]. Immature forms show a reduced nuclear:cytoplasmic ratio with larger, sometimes irregular, nuclei frequently with prominent nucleoli (Figure 16.3). Multinucleation, nuclear pseudo-inclusions (Dutcher bodies) and nuclear lobulations can be observed (Figure 16.4)[1]. Of note, immaturity and atypical nuclear features are usually not observed in reactive plasmacytosis and therefore are good indicators of malignancy. Plasmablasts can resemble immunoblasts with central, prominent eosinophilic nucleoli (Figure 16.5); sometimes myeloblast-like cells occur (Figure 16.6), and PCM with frankly anaplastic morphology and bizarre giant cells may raise a differential diagnosis of a non-haematopoietic neoplasm (Figure 16.7) [29, 30]. IgA-secreting PCM frequently shows large cells with broad, ‘flaming’ cytoplasm. Disturbances in Ig secretion can cause a variety of cytoplasmic changes, including Mott cells with multiple globular inclusions, eosinophilic Russell bodies with or without a discernible nucleus (Figure 16.8a, b), pseudo signet-ring cells (Figure 16.9) and rarely pseudo-Gaucher cells with fibrillary deposits or crystalline inclusions (Figure 16.10). Of note, these changes are not specific for malignancy and can be observed in reactive plasmacytosis to a minor degree. Rarely a crystal storing histiocytosis can accompany PCM [31, 32].
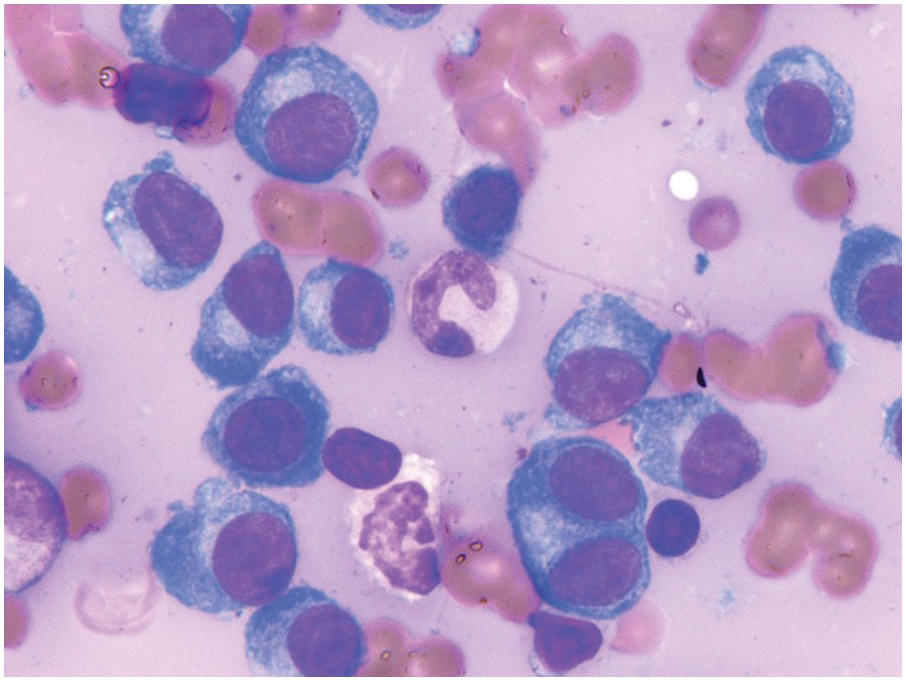
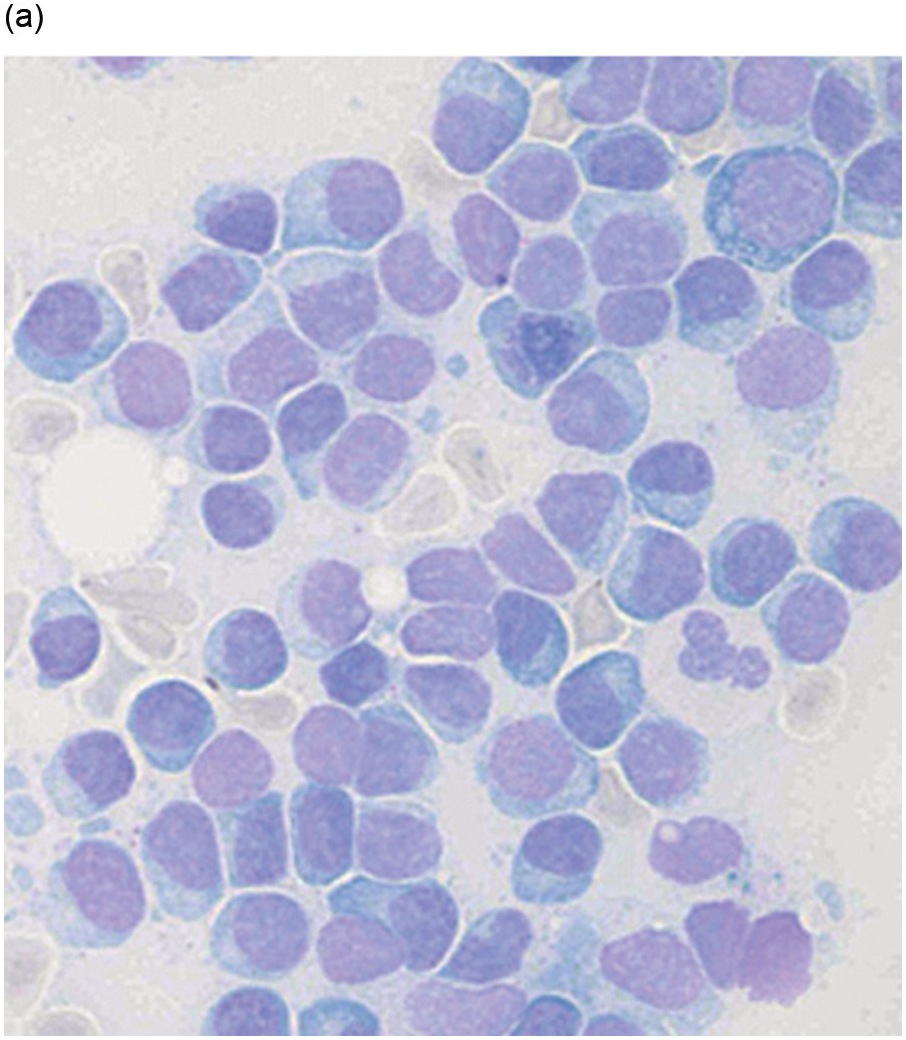
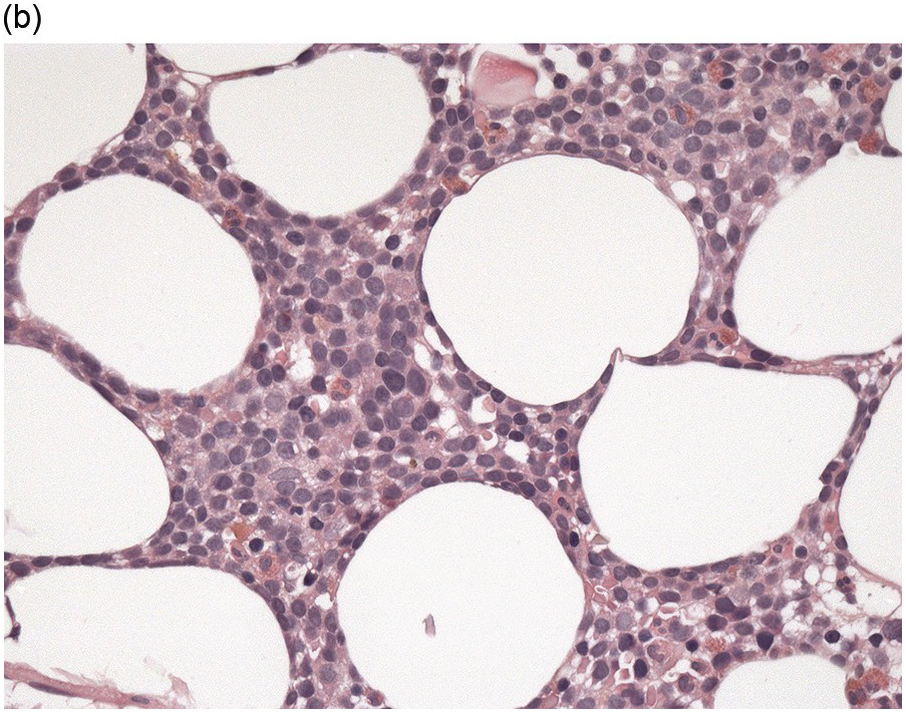
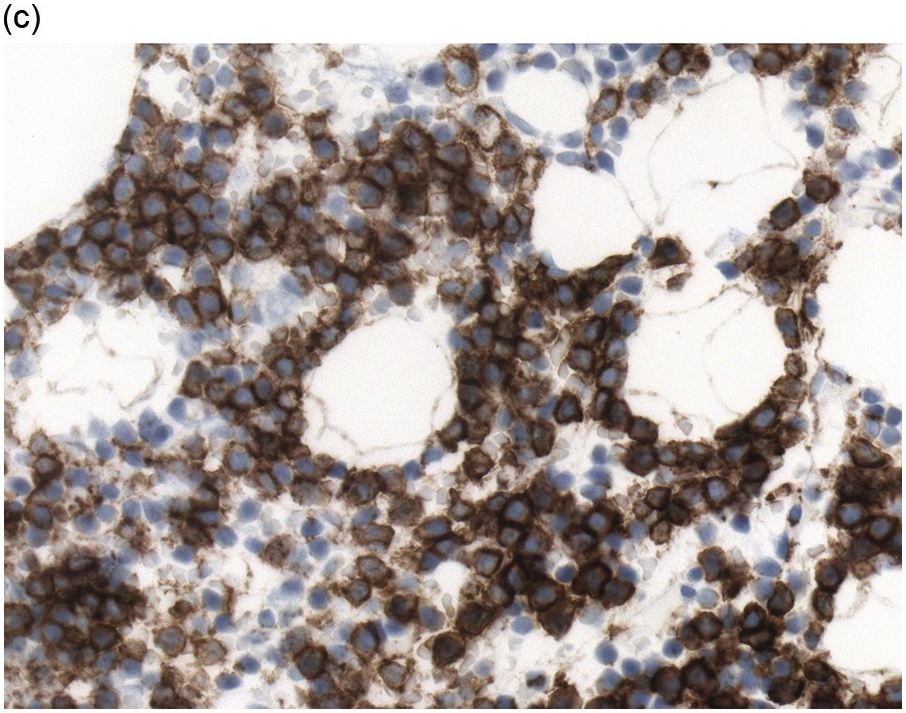
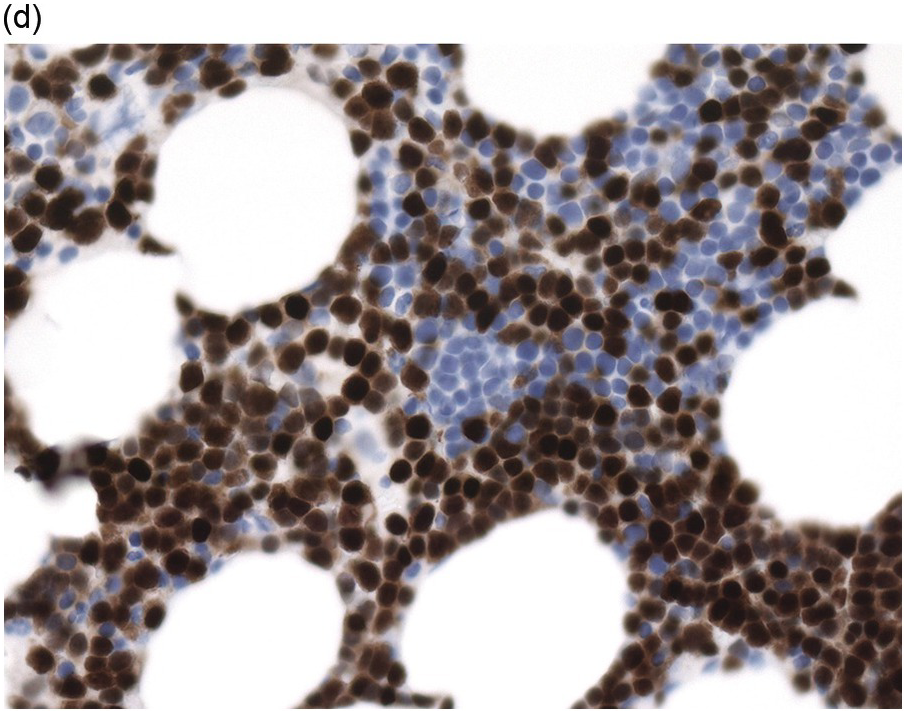
Figure 16.2 (a) Aspirate with neoplastic plasma cells with lymphoplasmacytic morphology with narrow cytoplasm with perinuclear hof and small nuclei. (b) Bone marrow biopsy with an interstitial infiltrate of plasma cells with lymphoplasmacytic morphology and strong expression of (c) CD20 and (d) Cyclin D1 as indication of the presence of translocation t(11;14).

(a) Plasma cell myeloma with interstitial infiltrate and multiple Russell bodies.
(b) Staining for kappa light chains also highlights interstitial distributed neoplastic plasma cells.
Although the nuclear features of PCM show a broad range, grading systems like the one proposed by Bartl et al. have lost their prognostic importance in comparison to clinical and cytogenetic parameters, but it is still relevant to highlight the plasmablastic subtype of PCM, which has a poor prognosis and an aggressive course [29, 30].
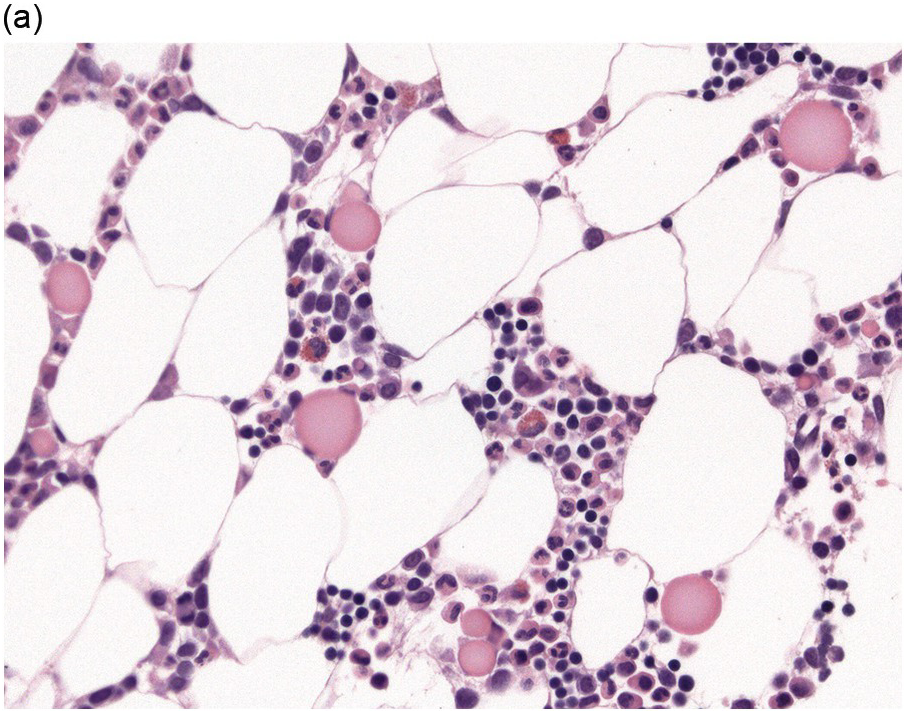
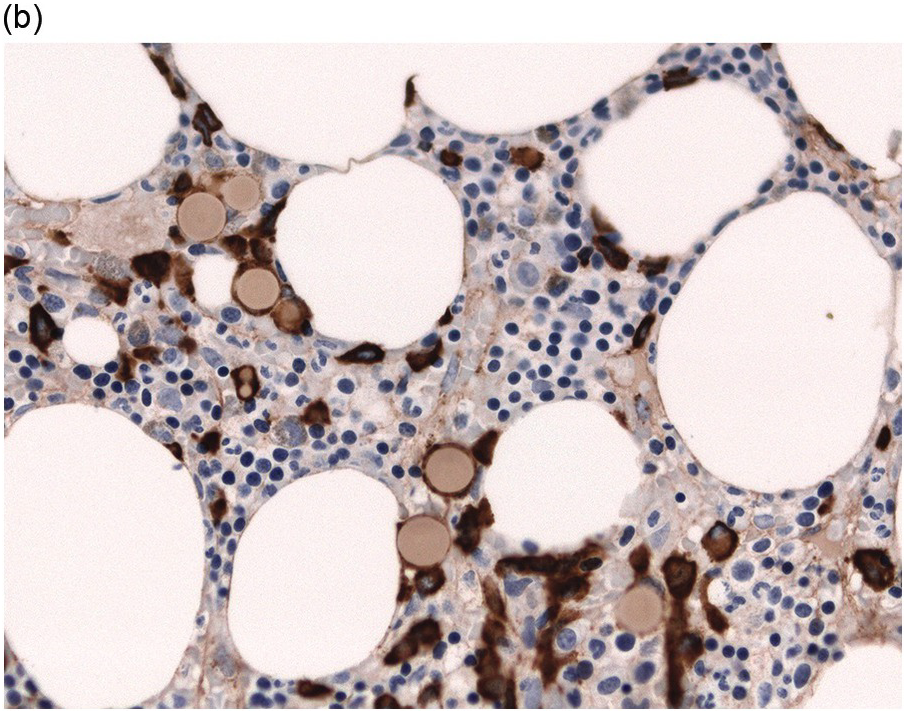
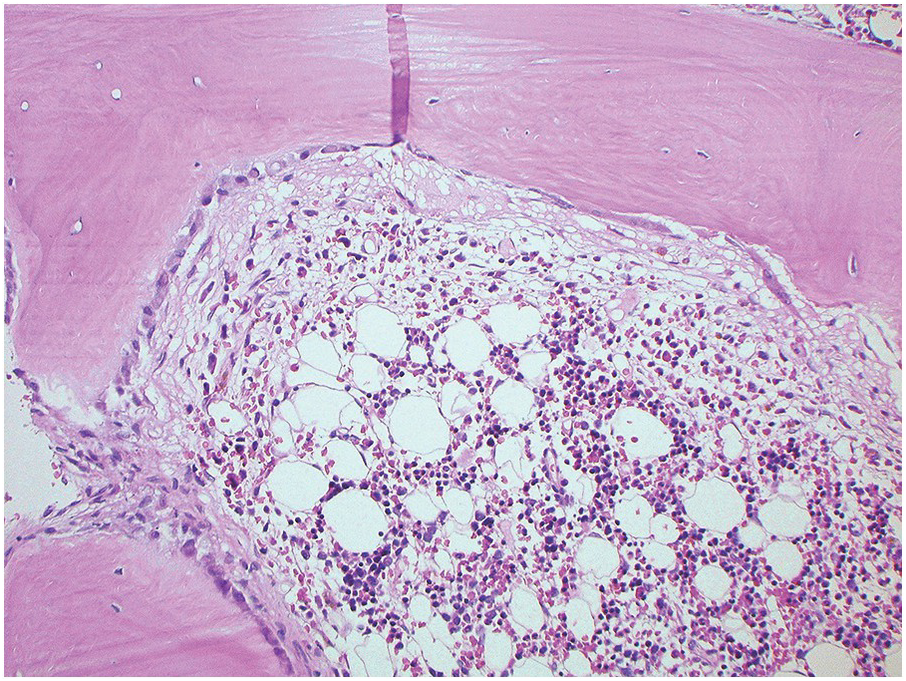
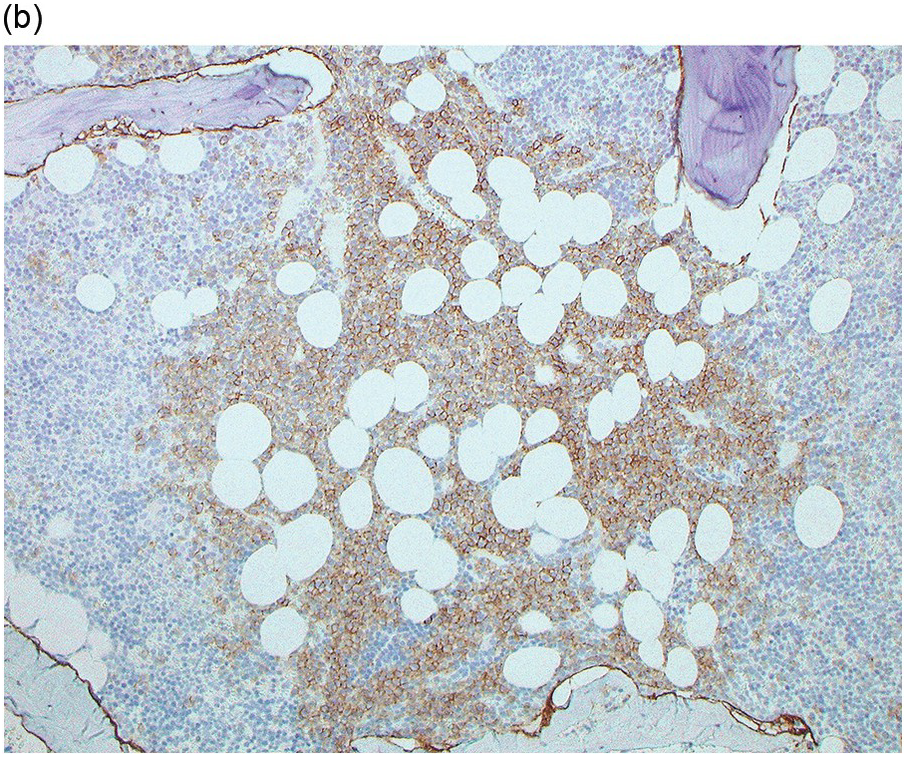
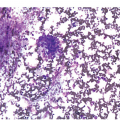
The BM biopsy is a mainstay for the diagnosis of PCM [29, 33]. In addition to cytological features, the distribution and arrangement of plasma cells is important for the distinction of reactive plasmacytosis from PCM. Clusters of plasma cells in the interstitium without connection to vessels and small nodular aggregates are highly suspicious for malignancy. Large nodular aggregates and sheet-like infiltrates, which can lead to the complete displacement of haematopoiesis are virtually diagnostic of PCM (Figure 16.3). If more than 30% plasma cells are present in the marrow, a diagnosis of PCM can be established irrespective of cytological atypia. Sometimes reactive changes and hyperplasia of haematopoiesis can obscure an interstitial infiltrate, and immunohistochemistry (IHC) is mandatory to assess the presence and extent of a plasma cell infiltrate. Interstitial infiltrates of small cell/lymphoplasmacytic PCM are especially easy to overlook (Figure 16.2b), and sometimes atypical plasma cells are hard to discriminate from immature erythropoietic cells (Figure 16.11a). In cases with focal, but atypical, infiltrates or significant cytologic atypia, the diagnosis can be established even with lesser infiltrates. Since infiltration frequently is very heterogeneously distributed, a biopsy of sufficient size is required. In cases with active bone disease, irregular contours of bone trabeculae and prominent osteoclastic activity are noted. Fibrosis occurs in up to 10% of cases, sometimes in peritrabecular fashion indicative of additional renal bone disease (Figure 16.12). Amyloidosis is observed in a subset of cases, its frequency depending on the method of investigation (see Chapter 3) [34].
(a) Hyperplastic haematopoietic marrow with an interstitial infiltrate of neoplastic plasma cells with occasional multinucleated cells (inset).
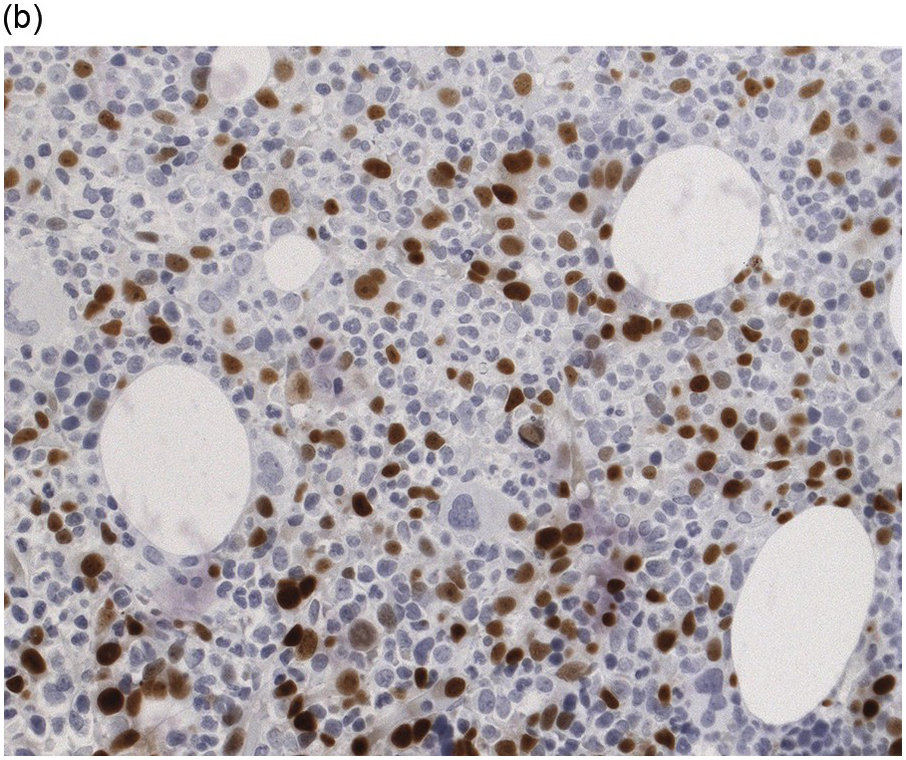
(b) MUM1 stain highlights the atypical nuclei of the interstitially distributed plasma cells.
Immunophenotype
Immunohistochemistry on the BM biopsy facilitates plasma cell quantification, highlights their distribution and allows identification of light chain restriction and an aberrant immunophenotype [33, 35, 36]. Furthermore, IHC, as well as flow cytometry, is very important in assessing residual disease after therapy. As mentioned above, small cell PCM with interstitial distribution can easily be overlooked in routine stains and requires IHC. Plasma cell myeloma shows the immunophenotype of normal plasma cells, with some important differences (Table 16.2). Of note, the frequencies of expression of any given marker quoted in the literature may vary depending of the mode of detection (IHC vs. flow cytometry). Due to the upregulation of the B-cell transcription factor IRF4/MUM1, terminally differentiated B-cells lose the expression of surface immunoglobulin and B-cell markers CD20 and CD22 and downregulate PAX5, whereas plasma cell antigens are expressed. In contrast to normal plasma cells, malignant plasma cells also lose CD19 in most cases, although this marker is of less diagnostic relevance in the BM biopsy. The most useful plasma cell markers are MUM1 (Figure 16.11b) and CD138 (syndecan) (Figure 16.13a), additional useful antigens are CD79a and CD38 [10, 33, 37–39]. Of note, none of these markers is specific for plasma cells; CD138 is expressed by many epithelial cells, CD79a and MUM1 by a subset of B-cells and CD38 by T-cells, though at lower intensity. PCM frequently shows no or only weak expression of CD45, which may lead to diagnostic problems, especially in cases with anaplastic cytology. CD56 (NCAM) is aberrantly expressed by >70% of PCM and can be used as a marker of malignancy (Figure 16.13b) [11]. Of note, clonal plasma cells in small B-cell lymphomas such as lymphoplasmacytic lymphoma are always CD56 negative [40]. Other surface markers expressed in a minority of PCM are CD117 (Figure 16.13c), CD20 and CD10, among others [28, 33, 41–44]. Approximately 20% of PCM express CD20, with strong and homogeneous positivity in a subset of cases [28]. Many of those exhibit lymphoplasmacytic morphology and may express other B-cell markers, such as PAX5 or CD22, but usually retain strong and homogeneous expression of CD138. Cyclin D1, which is absent from normal B-cells and plasma cells, is expressed in approximately 50% of PCM, with 20% showing strong and homogeneous nuclear expression due to the presence of the t(11;14) translocation (Figure 16.2c) [44, 45]. About half of the t(11;14)+ cases show lymphoplasmacytic morphology with frequent expression of CD20 (Figure 16.2d), which may lead to confusion with mantle cell lymphoma. MYC expression is common in PCM, unrelated to the presence or absence of MYC translocations, but is absent from MGUS and may be related to poor prognosis [46, 47]. Epithelial membrane antigen (EMA, MUC1) is expressed in the majority of cases, but also stains normal plasma cells [48].
Table 16.2 Immunohistochemical markers for plasma cell myeloma.
| Marker | Other relevant reactivity | Frequency in PCM | Remarks |
|---|---|---|---|
| CD138 (syndecan) | PC, epithelia | >95% | Reliable PC marker |
| MUM1/IRF4 | Terminally differentiated B-cells | >95% | Reliable PC marker |
| CD79a | Pan-B-cell, PC | >95% | Marks all B-cells |
| CD38 | Plasma cells, some B- and T-cells | >95% | Very strong reactivity in PC |
| CD19 | Pan-B-cell, 70% of normal PC | <5% | Loss as malignancy criterium in FCM |
| CD20 | Pan-B-cell | 10–20% | Variable, frequent in t(11;14)+ LP PCM |
| PAX5 | Pan-B-cell | Rare | Rare cases of t(11;14)+ PCM |
| CD45 | Pan leukocyte marker, variable in normal PC | 20–30%, frequently weak | May result in misdiagnosis as non-haematopoietic neoplasm |
| CD56 (NCAM) | NK cells, very rare normal PC | 70–80% | Infrequent and weak in MGUS |
| Cytoplasmic immunoglobulins | IgG >50%; IgA 20% Light chains 15–20% IgD, IgE, IgM rare | Intensity frequently less than in normal PC Rare biclonal PCM 3% nonsecretory (may be positive by IHC) | |
| Cyclin D1 | Non-lymphoid cells | 40–50%, also in t(11;14)+ MGUS | Strong and homogeneous in t(11;14)+ cases (20%), 20-30% heterogeneous (frequently trisomy 11) |
| CD117 | Mast cells, myeloblasts | 20–35% | Favourable prognosis |
| EMA | Epithelial cells, normal PC | 60–90% | Also expressed by some plasmablastic lymphomas |
PC: plasma cell; PCM: plasma cell myeloma; FCM: flow cytometry; IHC: immunohistochemistry; MGUS: monoclonal gammopathy of unknown significance; EMA: epithelial membrane antigen.
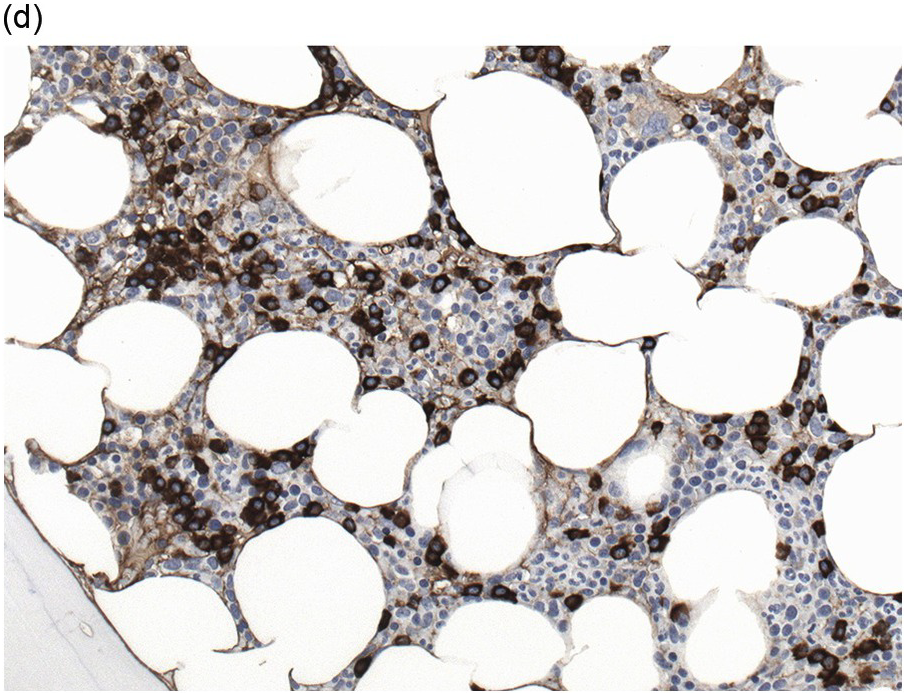
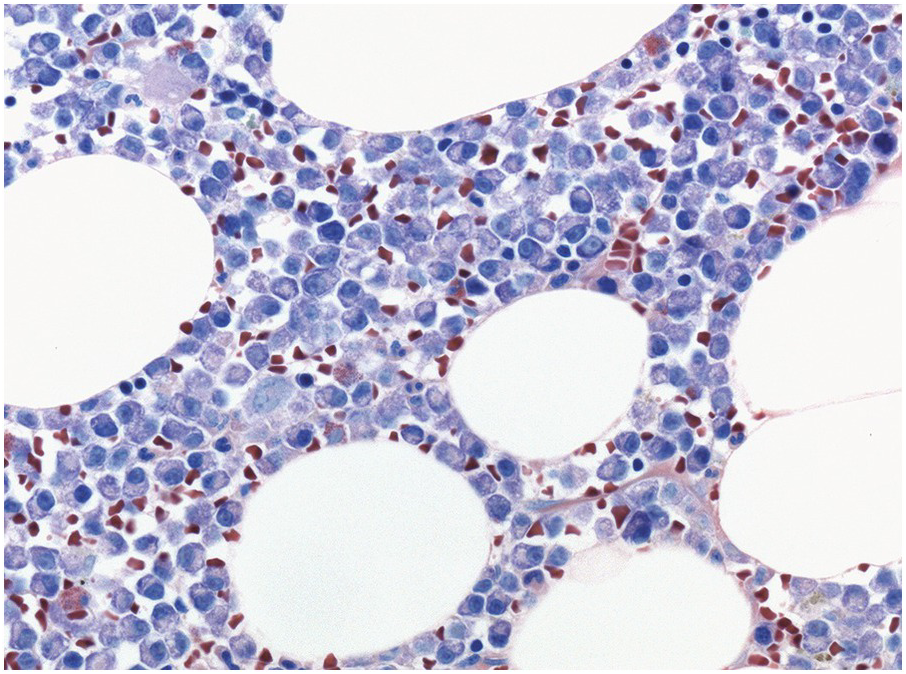
Stains for immunoglobulin light chains are essential to confirm a monotypic plasma cell population (Figures 16.8b, 16.13d). Light chain ratios of 8:1 up to 16:1 and higher have been proposed to distinguish MGUS from PCM [49]. Since immunoglobulin expression is frequently much lower in neoplastic as compared to reactive plasma cells, and plasma insudation can create significant background staining, demonstration of light chain restriction can be difficult by IHC. Alternatively, colourimetric in situ hybridization (CISH) for kappa and lambda light chains is a very robust method and has the advantage of lacking the background staining caused by serum immunoglobulins. CISH can be employed as double staining, further facilitating the determination of light chain restriction [50]. Staining for heavy chains can be used in addition, keeping in mind that 20% of PCM produce light chains only. As mentioned above, IgA-producing PCM frequently shows large irregular plasma cells with ‘flaming’ cytoplasm. IgM-positive PCM, which does not undergo heavy chain class switch, is rare, making up less than 1% of cases. It shows conventional morphology, homogeneously expresses plasma cell markers, usually lacks CD 56 and CD117 and is frequently positive for cyclin D1 due to presence of the t(11;14) translocation [51]. A pitfall in the assessment of immunoglobulin expression is the rare occurrence of biclonal PCM with production of different heavy chains or light chains. In addition, rare cases of PCM with production of both light chains in the same cell, usually with secretion of only one light chain, have been observed [52].
Flow cytometry is extensively used for staging and follow-up of PCM and may help to identify aberrant immunophenotypes, as well as minimal residual disease [9, 53, 54].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


