Abstract
Neoplasms arising from precursor lymphoid cells committed to the B-cell or T-cell lineage can present primarily in the bone marrow (BM), blood (i.e. leukaemic presentation) or at extramedullary tissue sites (i.e. lymphomatous presentation) (Table 14.1). Hence, these neoplasms are appropriately termed as B- or T-lymphoblastic leukaemia/lymphoma [1, 2].
Introduction
Neoplasms arising from precursor lymphoid cells committed to the B-cell or T-cell lineage can present primarily in the bone marrow (BM), blood (i.e. leukaemic presentation) or at extramedullary tissue sites (i.e. lymphomatous presentation) (Table 14.1). Hence, these neoplasms are appropriately termed as B- or T-lymphoblastic leukaemia/lymphoma [1, 2].
Table 14.1 Summary of the various entities precursor lymphoid neoplasms accepted in the WHO 2017 revisions.
1. B-lymphoblastic leukaemia/lymphoma
a. B-lymphoblastic leukaemia/lymphoma, not otherwise specified (NOS)
b. B-lymphoblastic leukaemia/lymphoma with recurrent genetic abnormalities
i. B-lymphoblastic leukaemia/lymphoma with t(9;22)(q34;q11.2); BCR-ABL1
ii. B-lymphoblastic leukaemia/lymphoma with t(v;11q23.3); KMT2Aa-rearranged
iii. B-lymphoblastic leukaemia/lymphoma with t(12;21)(p13.2;q22.1);ETV6-RUNX1
iv. B-lymphoblastic leukaemia/lymphoma with hyperdiploidy
v. B-lymphoblastic leukaemia/lymphoma with hypodiploidy
vi. B-lymphoblastic leukaemia/lymphoma with t(5;14)(q31.1;q32.1);IGH/IL3
vii. B-lymphoblastic leukaemia/lymphoma with t(1;19)(q23;p13.3);TCF3-PBX1
viii. bB-lymphoblastic leukaemia/lymphoma, BCR-ABL1‑like
ix. bB-lymphoblastic leukaemia/lymphoma with iAMP21
2. T-lymphoblastic leukaemia/lymphoma
3. NK-lymphoblastic leukaemia/lymphoma
a Gene named changed per convention from MLL to KMT2A. Multiple rearrangement partners are recognized and hence the ‘v’ designation in the translocation.
b Newly recognized entities not previously listed in the WHO 2008 [3].
Synonyms
Precursor B-cell acute lymphoblastic leukaemia; pro-B-lymphoblastic leukaemia; pre-B-lymphoblastic leukaemia; B-cell lymphoblastic lymphoma; T-lymphoblastic leukaemia; precursor T-cell acute lymphoblastic leukaemia.
Epidemiology
Although precursor lymphoid neoplasms only comprise about 0.4% of all new cancers (in adult and paediatric age groups), 56.1% of new cases of acute lymphoblastic leukaemia (ALL) occur in patients less than 20 years old with 85% of these representing B-ALL [4]. However, among lymphoblastic lymphoma (LBL) cases, only 10% are of B-cell origin while most are of T-cell origin [5]. The incidence rates are higher in Hispanic and Caucasian populations compared to other races/ethnicities. For treatment purposes, it must be recognized that the National Comprehensive Cancer Network (NCCN) recognizes the adolescent and young adults (AYA) definition of the National Cancer Institutes (NCI) as patients aged between 15 and 39 years and note the better outcome of this population based on paediatric treatment protocols [6].
Clinical Presentation
Most patients diagnosed with B-ALL present initially with symptoms related to BM infiltration (bone pain and arthralgias) and resulting cytopaenias such as fatigue, malaise (due to anaemia), infections (neutropaenia) and bleeding/ecchymoses (due to thrombocytopaenia). Extramedullary infiltration may also been seen variably in B-ALL often in the form of lymphadenopathy. Cutaneous involvement is also seen relatively frequently in B-ALL/LBL (often in the head and neck region) compared to T-ALL/LBL where presentation as mediastinal masses with effusions is much more common [7]. The peripheral blood (PB) white blood cell (WBC) counts can be very variable including leukopaenia (even aleukaemic), normal with a few circulating blasts to cases with very high WBC counts and numerous blasts. Isolated lymphomatous presentation without leukaemic component of B-LBL is uncommon but has been described [8, 9]. About 10% of cases may involve sanctuary sites (CNS or testes).
Morphologic Diagnosis
Peripheral Blood
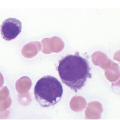
Nearly half the patients have a WBC count less than 10,000/μL and numbers of circulating blasts may be variable. Hyperleukocytosis (WBC >50,000/μL) is a feature seen more frequently in patients with T-ALL and is a negative prognostic indicator. Blasts exhibit lymphoid cytomorphology and are variably sized, generally one to two times larger than mature lymphocytes. It must be noted that circulating lymphocytes in infants often look a little immature compared to lymphocytes in older children (Figure 14.1a, b) and flow cytometry is helpful in excluding the possibility of leukaemia in suspicious cases. Lymphoblasts typically exhibit fine nuclear chromatin with a variable number of inconspicuous nucleoli on Wright–Giemsa smears (Figure 14.2). Cytoplasm is scant but may occasionally be basophilic, vacuolated or even rarely exhibit fine granulations without Auer rods and the presence of granules does not necessarily connote myeloid lineage.
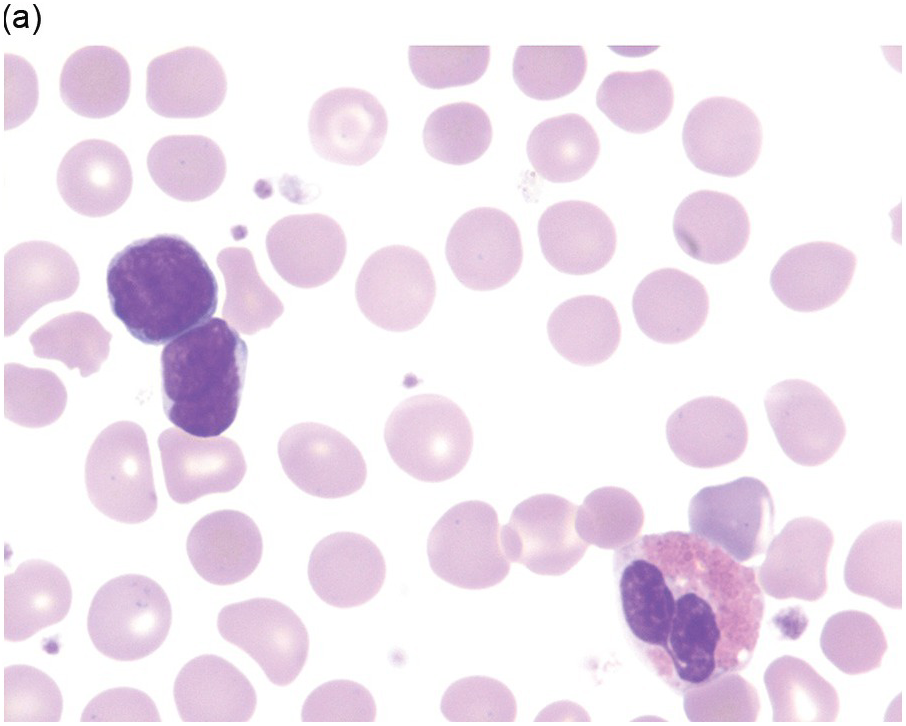
(a) Low power depicting a neonate with alloimmune neutropaenia and several peripheral blood small lymphoid cells.
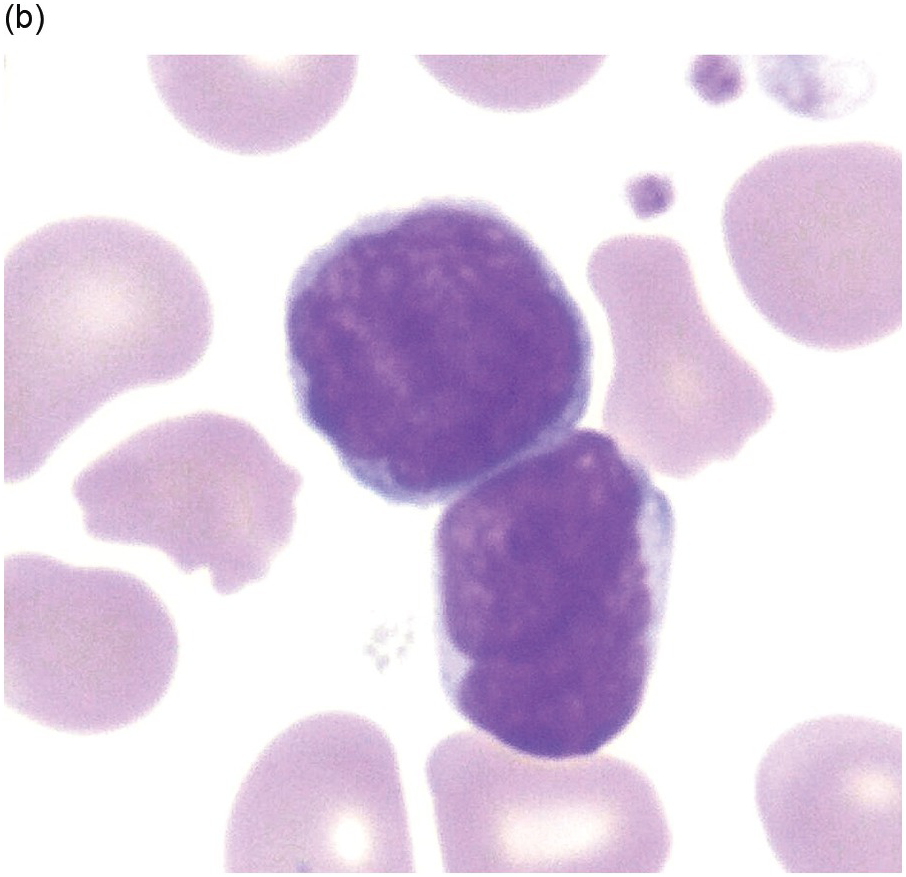
(b) Higher power shows cells with blastoid cytomorphology and nuclear convolutions/indentations and increased nuclear cytoplasmic ratio. On closer inspection, the cells exhibit a spectrum of cell size including some forms that are mostly the size of normal large lymphocytes. Flow cytometric analysis confirmed the predominant peripheral blood lymphoid population to be reactive T-cells. (Wright–Giemsa)
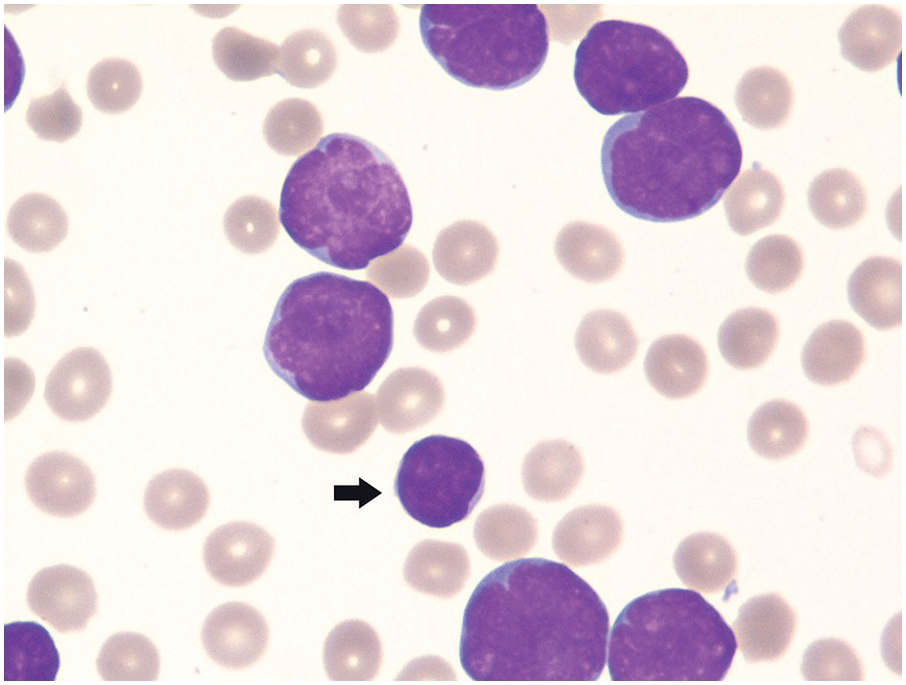
Figure 14.2 Circulating lymphoblasts in precursor B-cell acute lymphoblastic leukaemia. Blasts are medium to large in size in comparison with the small lymphocyte (arrow). The blasts exhibit high NC ratios and variably prominent nucleoli. Also note the absence of platelets in the background, in keeping with concurrent thrombocytopaenia. (Wright–Giemsa)
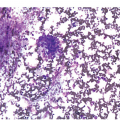
Classification systems were developed earlier based on blast cytomorphology (size, clefting and nucleoli) but are not used for prognostication currently [10]. Cases with underlying hyperdiploidy at the genetic level often exhibit large blasts three to four times the size with a folded nuclear membrane and variably prominent nucleoli. Prior to performing flow cytometry and BM biopsy (BMB), useful things to assess include lack of dysplasia in neutrophils and absence of Auer rods in blasts (as may be seen in AML with t(8;21)). Rarely, striking eosinophilia can be present and some of these cases are often oligoblastic and may be of B- or T-cell lineage, either of which could harbour an underlying t(5;14) translocation (but with different breakpoints in each). Concurrent anaemia and thrombocytopaenia are often present in ALL. Lastly, circulating reactive large mononucleosis (T-) cells in the setting of primary infectious mononucleosis may be flagged as lymphoblasts by the laboratory technicians and hence knowledge of the clinical setting is important (Figure 14.3). In difficult cases, flow cytometry will help in confirming that the circulating atypical cells are reactive CD8 T-cells.
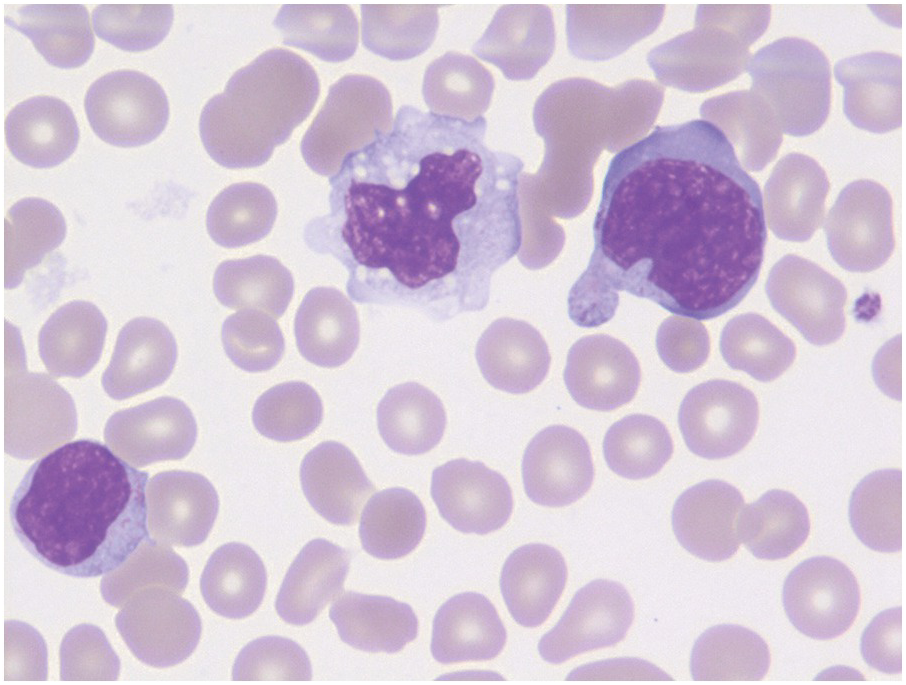
Figure 14.3 Large atypical reactive T-cells in infectious mononucleosis. The pleomorphic and variable size with deep-blue cytoplasm allows for distinction from blastic cells morphologically. A normal monocyte is detected in the centre of the field for comparison. (Wright–Giemsa)
Bone Marrow
Core Biopsy
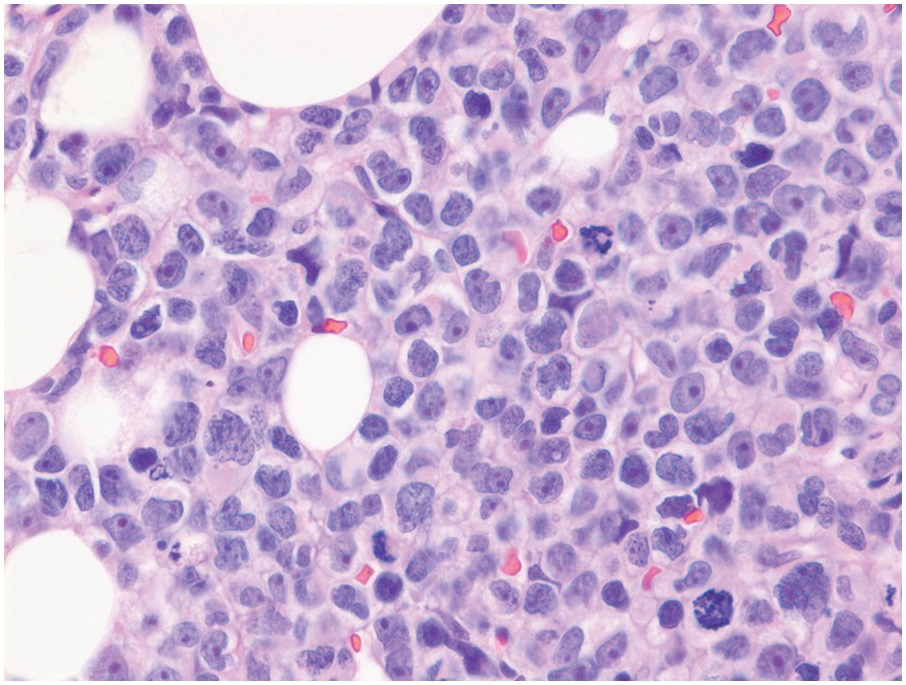
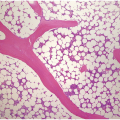
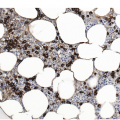
The BMB is useful in characterizing the extent of BM involvement and most cases demonstrate high cellularity approaching 100% with replacement by blastic leukaemic cells throughout the interstitium with only scattered preserved megakaryocytes in between (Figure 14.4). Cases with diffuse involvement and high proliferation often impart a ‘starry sky’ cytomorphology akin to Burkitt lymphoma. However, the extent of involvement can be variable with patchy interstitial involvement intermingled with substantial amounts of residual normal haematopoietic cells. Extensive necrosis may rarely be seen [11] (Figure 14.5) in addition to striking eosinophilia especially in cases of B-ALL with underlying t(5;14). On H&E sections, the blasts exhibit back-to-back nuclei with scant cytoplasm with finely dispersed nuclear chromatin and inconspicuous nucleoli. Brisk mitotic activity is also readily appreciated (Figure 14.6). Rarely, extensive fibrosis may be responsible for a dry tap. B-ALL subsets with high-hyperdiploidy were reported more frequently to harbour reticulin fibrosis at diagnosis although there was no prognostic significance for fibrosis [12].
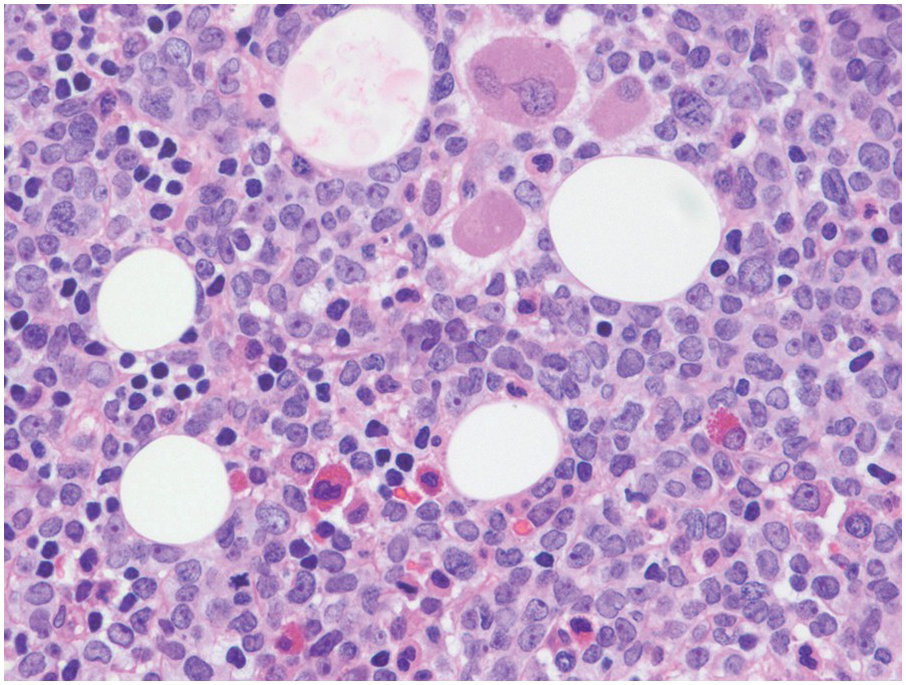
Figure 14.4 Precursor B-ALL extensively involving the bone marrow biopsy. Blasts are medium sized and replace most of the marrow spaces with very little residual megakaryocytes (on the top) and few scattered residual erythroid precursors and eosinophils. (H&E)
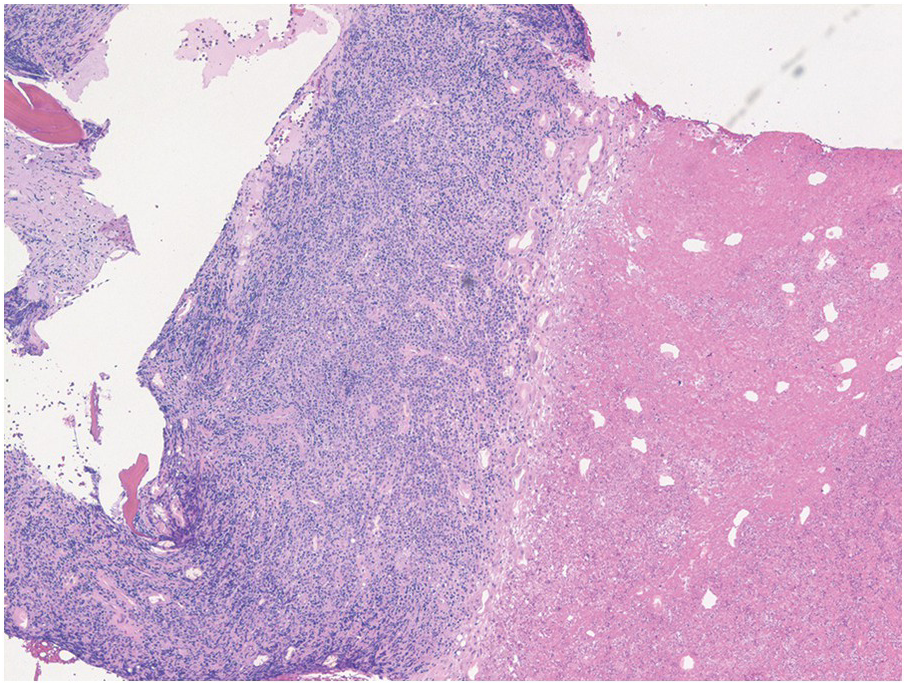
Figure 14.5 Precursor B-ALL involving bone marrow core biopsy with extensive infarction/necrosis noted on the right side. (H&E)
Aspirate Smears
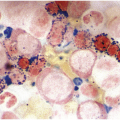
On BM aspirate smears, spicules are typically hypercellular with similar morphology to blasts on the peripheral smear (Figure 14.7). In cases with a packed BM and a dry tap, touch imprints are useful in assessing blast cytomorphology. Granular blasts with azurophilic granules and forms with hand-mirror cytomorphology may be seen although there is no prognostic significance to these morphologic findings. Cytochemistry for myeloperoxidase can help in confirming the lack of myeloid differentiation in such cases (Figure 14.8). In cases with few blasts, distinction from reactive haematogones (B-cell precursors; Figure 14.9) can be difficult, although with careful examination it can be discerned that these cells have a spectrum of cell size between blasts and mature lymphocytes reflective of their progressive maturation to mature naïve B-cells.
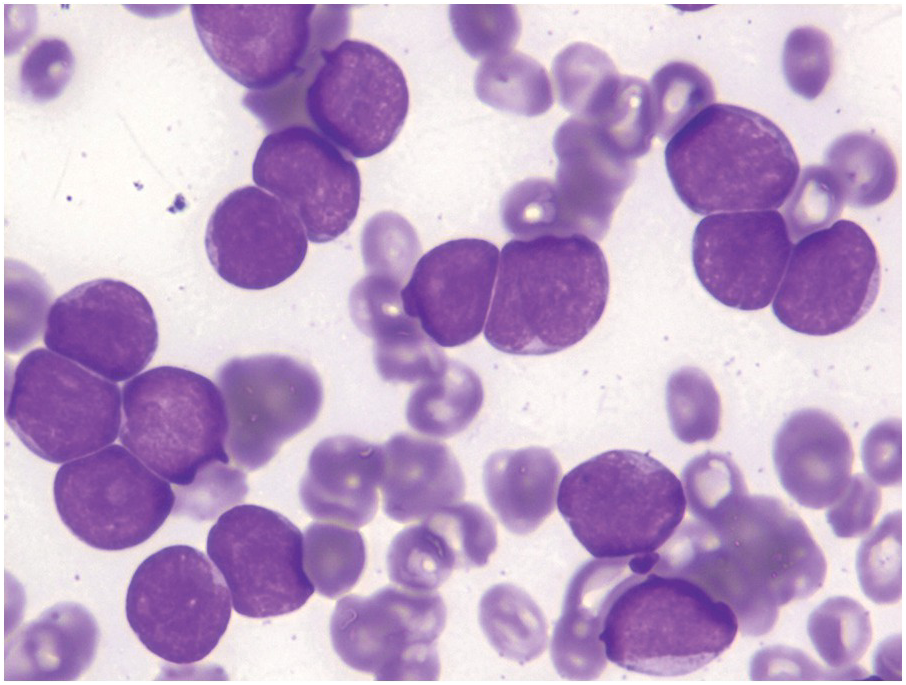
Figure 14.7 Bone marrow aspirate smear blasts in B-ALL demonstrating identical cytomorphology as noted in the peripheral blood blasts depicted in Figure 14.2. (Wright–Giemsa)
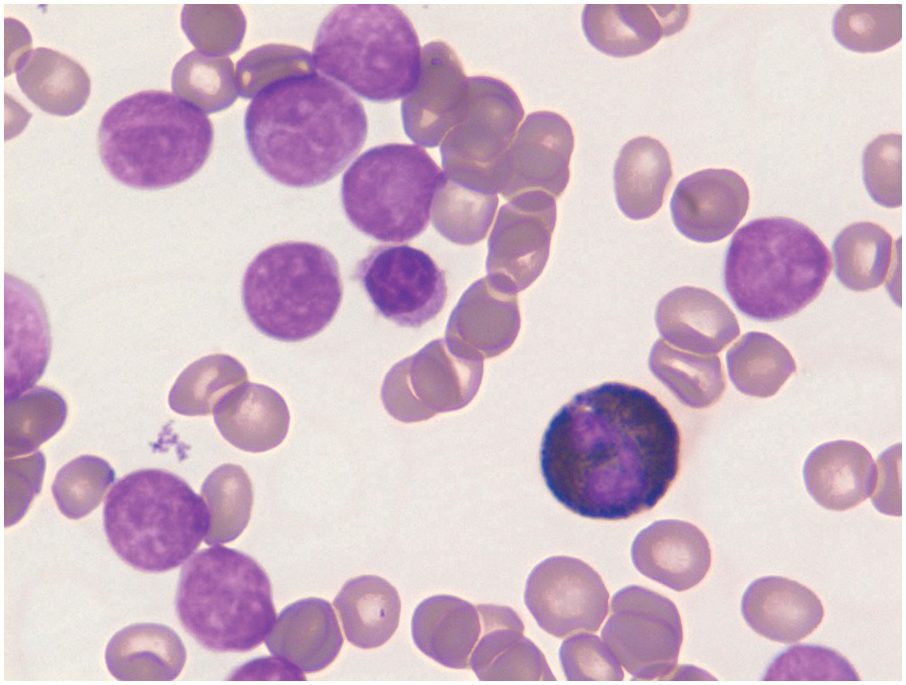
Figure 14.8 Cytochemistry for myeloperoxidase showing B-ALL blasts negative for cMPO while a neutrophil in the centre is strongly positive. (Myeloperoxidase cytochemistry)
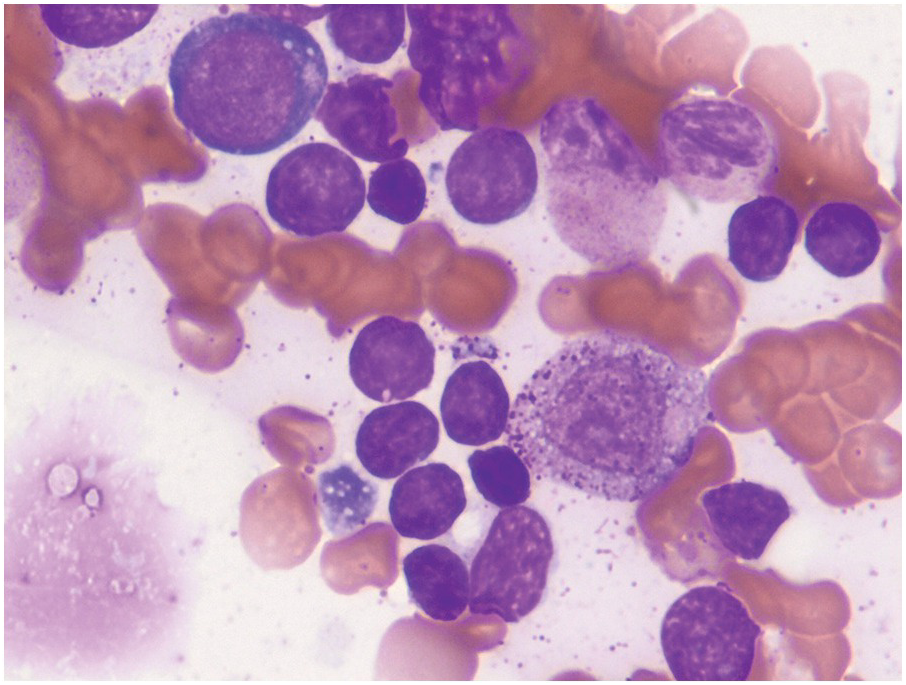
Figure 14.9 Bone marrow haematogones on aspirate smear of a regenerative bone marrow after chemotherapy with numerous small to medium-sized lymphoid cells compatible with haematogones. These cells have very scant cytoplasm with a spectrum in cell size consistent with their progressive maturation to mature naïve B-cells. (Wright–Giemsa)
Ancillary Diagnostic Techniques
Consideration of Normal B- and T-Cell Development
A brief knowledge of the stages of development of B- and T-cells is essential for understanding the different subsets of B- and T-ALL. Some of the subsets recapitulate expression patterns of antigens appropriate for the normal developmental stage.
B-Cell Development
Normal B-cell development starts in the BM with origin from a haematopoietic stem cell (HSC) that differentiates into a common lymphoid progenitor (CLP). The CLP in turn, under the influence of certain transcription factors such as E2A, early B-cell factor (EBF1) and PAX5 results in a committed B-cell precursor called the early ‘pro-B’ cell [13]. Beyond this stage, PAX5 plays a critical role in maintaining a B-cell identity all the way through the evolution of a mature naïve B-cell with functional surface light chain expression, which exits the BM to nodal post-germinal B-cells until before the plasma cell stage when there is downregulation of PAX5 [13, 14]. Thus, all B-ALL subtypes arise from PAX5+B-cells that are arrested at various stages of development preceding the surface light expressing mature naïve B-cell.
T-Cell Development
The earliest T-cell progenitors originate from the BM [15] and migrate to the thymus where they evolve into CD4/CD8 double negative progenitors and undergo ‘education’ with resulting development of the various T-cell subsets including the CD4, CD8 single positive as well as the gamma-delta T-cell subsets. NOTCH is an important factor in the commitment and development of the T-cell lineage analogous to PAX5 for B-lineage commitment and development.
Immunophenotype
B-ALL
In most cases with circulating blasts and good BM aspirates, flow cytometry is critical for establishing baseline phenotypic signature of the blasts. Immunohistochemistry is useful when demonstration of TdT or cytoplasmic markers by flow cytometry is technically challenging. Demonstration of typical B-lineage markers (CD19, cCD22, cCD79a) in conjunction of markers of immaturity such as CD34 and/or TdT (Figures 14.10 and 14.11) allow establishment of the diagnosis of B-ALL/LBL. Lack of cCD3 and cMPO is also critical to exclude mixed phenotype acute leukaemia although these are very uncommon in the paediatric age group. However, expression of certain myeloid-associated antigens such as CD13 and/or CD33 is frequently present and does not warrant a designation as mixed phenotype leukaemia with B- and myeloid derivation. Further subclassification can be performed based on the maturational stages as:
1. Pro-B type (CD10–/TdT+)
2. Common type (CD10+) or
3. Pre-B (cIgM+/CD34–) types reflective of the normal maturational stages.
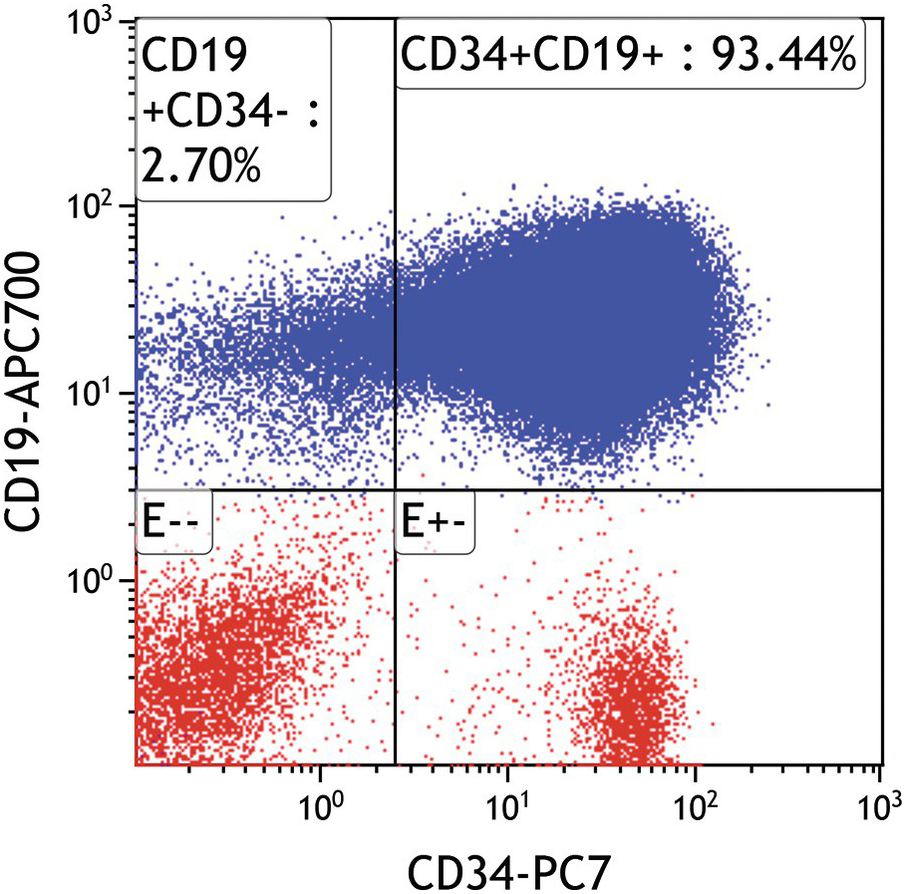
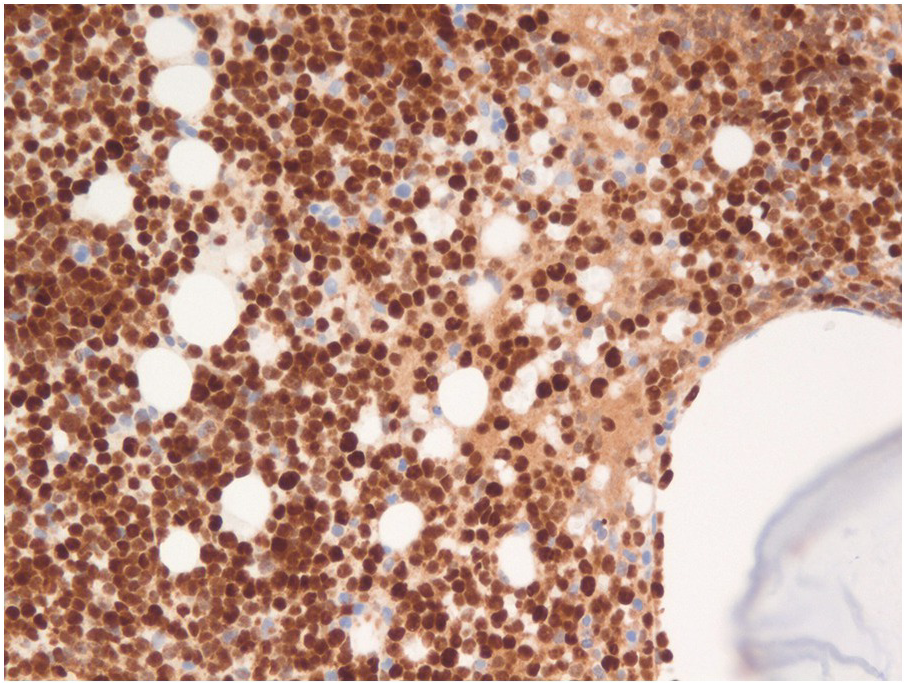
Figure 14.11 Precursor B-ALL expressing strong nuclear TdT expression by immunohistochemistry on the bone core biopsy. Both B- and T-ALL, as well as precursor myeloid malignancies to a lesser extent, express TdT, so TdT expression does not indicate any lineage differentiation. (TdT immunoperoxidase)
Asynchronous, uniform and/or aberrant expression of several markers is characteristic of B-lymphoblasts. While haematogones express CD10 (as do cases of ‘common type’ B-ALL), distinction from B-ALL is fairly easy by flow cytometry (Figure 14.12a–d) since haematogones exhibit a very characteristic maturation pattern with early haematogones expressing CD34 without CD20 [16] while late stage III haematogones lose CD34 and CD10 with progressive acquisition of CD20 [17]. Surface light chain expression is typically absent in most cases of B-ALL. However, transitional cases with restricted surface light chain and/or TdT with MYC and BCL2 alterations (double hit genetics) have been reported in rare instances and distinction from blastoid lymphoma/Burkitt leukaemia is sometimes difficult in these cases [18, 19]. Assessment of CD19 and cCD22/sCD22 expression on blasts is critical in relapsed disease since targeted therapy is available for both antigens (blinatumomab and inotuzumab, respectively).
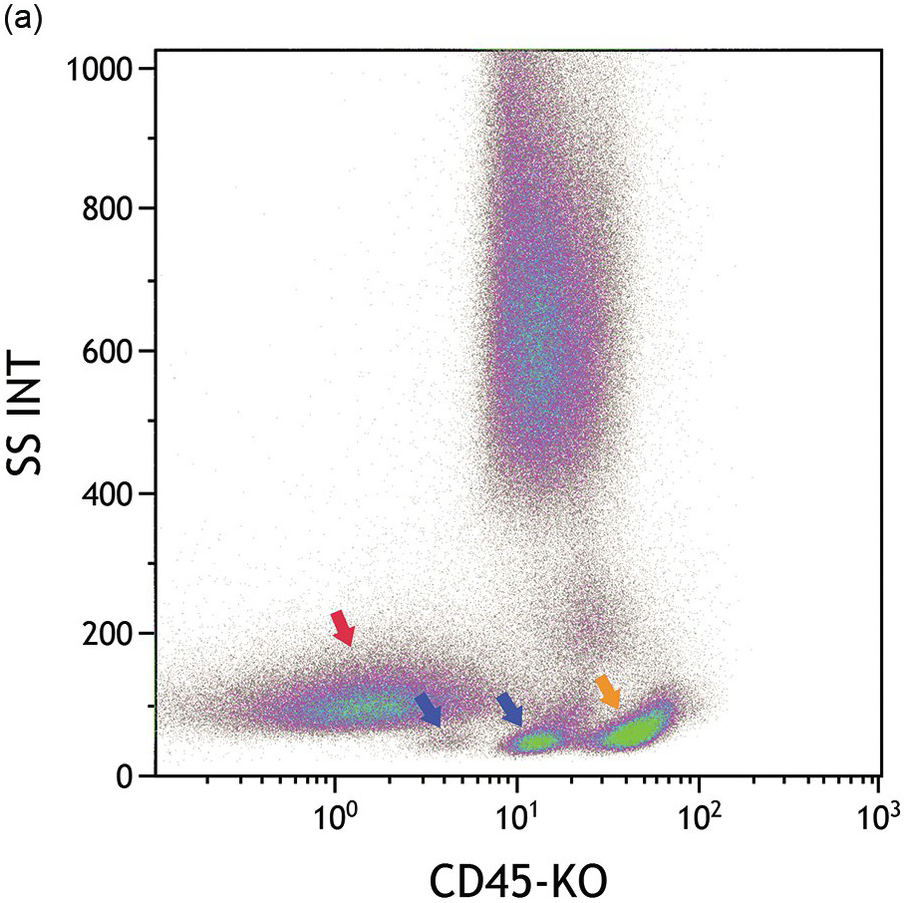
(a) CD45/side scatter plot shows three distinct populations with low side scatter including blasts (red arrow) with very dim CD45, and haematogones with dim CD45 (dual blue arrows) and mature lymphocytes (orange arrow) with moderate CD45 expression. Each of these populations are coloured accordingly in the other plots.
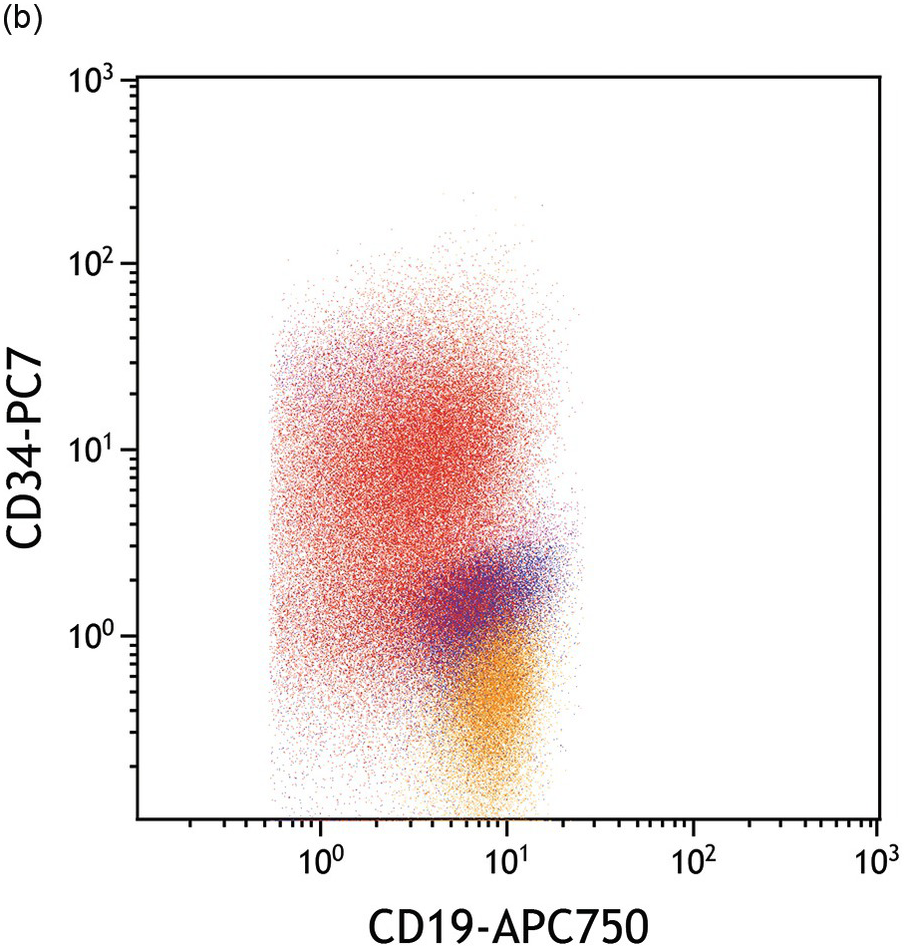
(b) Blasts express bright CD34 relative to the haematogones.
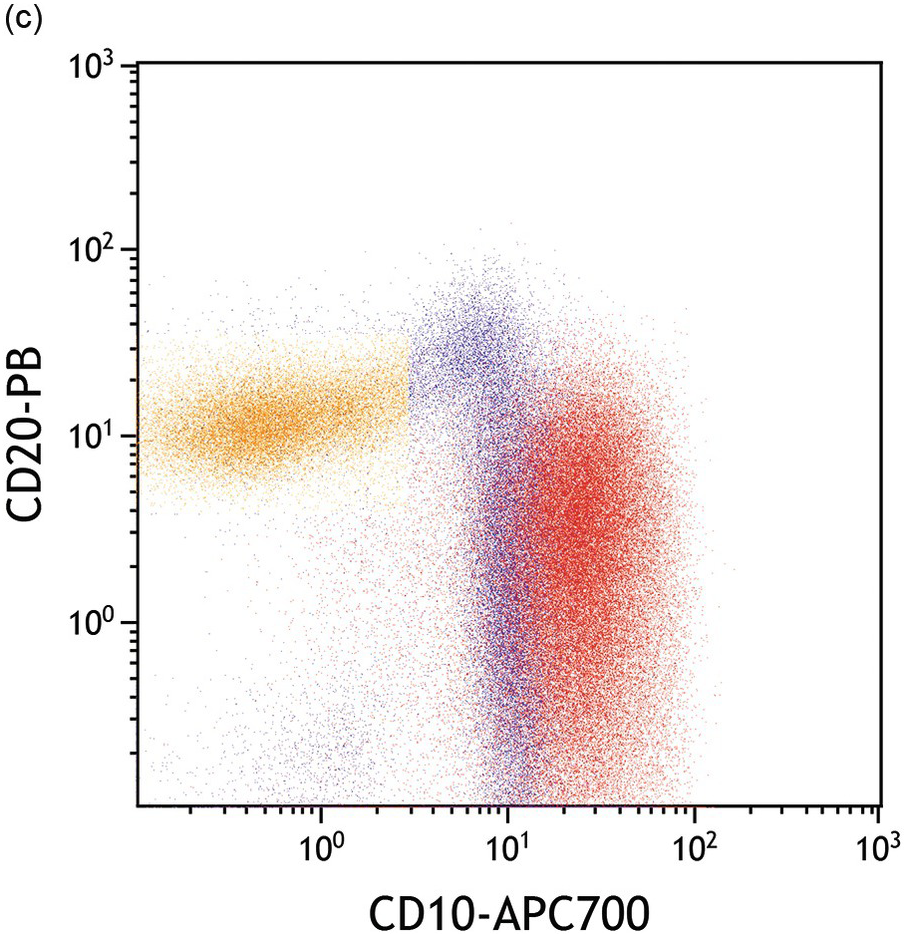
(c) Blasts exhibit abnormal bright CD10 relative to haematogones while the maturation spectrum of haematogones CD10 bright/CD20-negative (immature) to CD20+/CD10– mature B-cells is apparent in this plot.
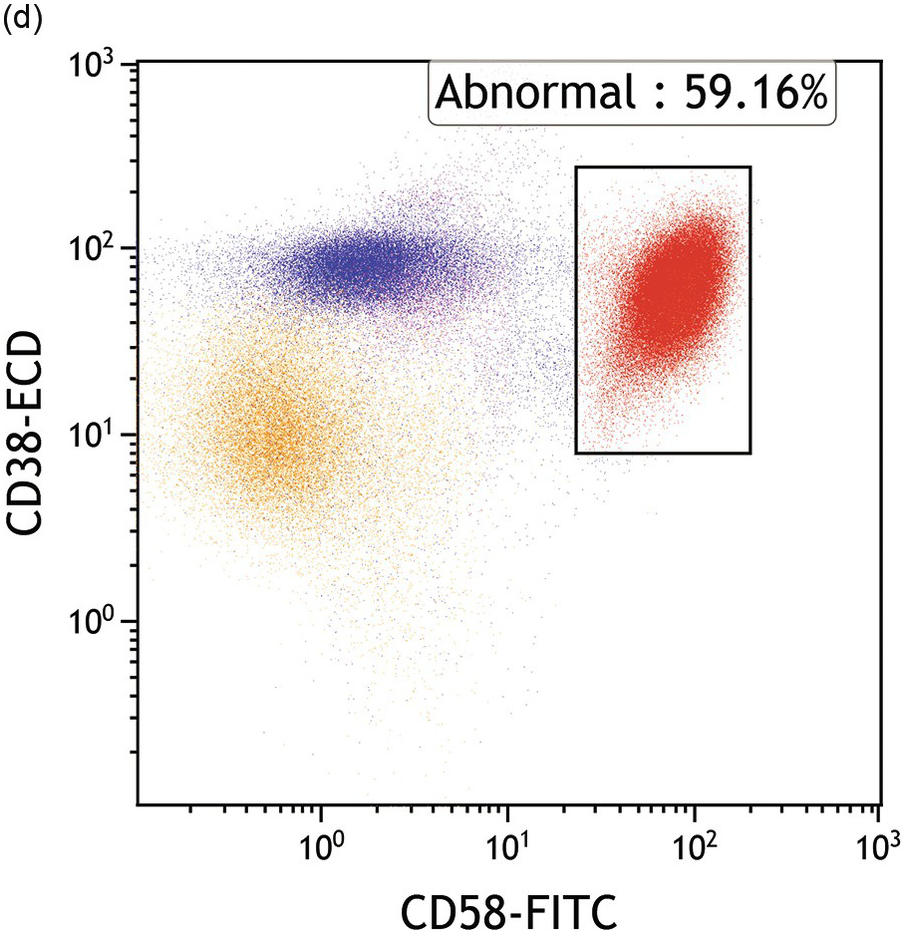
(d) Blasts express abnormal bright CD58 while both blasts and haematogones express moderate-bright CD38.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


