Abstract
Myeloid and lymphoid neoplasms with eosinophilia (MLNE) and rearrangements of PDGFRA, PDGFRB and FGFR1 were recognized as a standalone category in the 2008 WHO classification. PCM1-JAK2 was added to this family as a new provisional entity in the 2016 WHO classification [1, 2]. The features shared by neoplasms in this category include a common presentation with eosinophilia or hypereosinophilia in peripheral blood and an increased number of eosinophilic forms in bone marrow (BM). Some cases present as acute leukaemia. Some cases may lack hypereosinophilia. The underlying mechanism is the overexpression of an aberrant tyrosine kinase as a result of a fusion gene, or rarely of a mutation, and a diagnosis and classification requires the demonstration of the specific gene fusions. The cell of origin is a mutated pluripotent stem cell that has the potential to involve myeloid, lymphoid or both lineages, concomitantly or sequentially, leading to clinically complex and heterogeneous manifestations. A common scenario is the presentation as a chronic myeloproliferative neoplasm (MPN), usually with eosinophilia followed within a variable time period and depending on the gene fusion involved, by a progression to acute myeloid leukaemia (AML) or mixed phenotype acute leukaemia (usually in the BM), and B- or T-lymphoblastic leukaemia/lymphoma (B-/T-ALL) in BM or in an extramedullary site. Thus it is critical to recognize the clinicopathologic features of these neoplasms, identify the molecular genetic lesions and classify them accordingly. An accurate diagnosis and classification have important therapeutic and prognostic implications.
Introduction
Myeloid and lymphoid neoplasms with eosinophilia (MLNE) and rearrangements of PDGFRA, PDGFRB and FGFR1 were recognized as a standalone category in the 2008 WHO classification. PCM1-JAK2 was added to this family as a new provisional entity in the 2016 WHO classification [1, 2]. The features shared by neoplasms in this category include a common presentation with eosinophilia or hypereosinophilia in peripheral blood and an increased number of eosinophilic forms in bone marrow (BM). Some cases present as acute leukaemia. Some cases may lack hypereosinophilia. The underlying mechanism is the overexpression of an aberrant tyrosine kinase as a result of a fusion gene, or rarely of a mutation, and a diagnosis and classification requires the demonstration of the specific gene fusions. The cell of origin is a mutated pluripotent stem cell that has the potential to involve myeloid, lymphoid or both lineages, concomitantly or sequentially, leading to clinically complex and heterogeneous manifestations. A common scenario is the presentation as a chronic myeloproliferative neoplasm (MPN), usually with eosinophilia followed within a variable time period and depending on the gene fusion involved, by a progression to acute myeloid leukaemia (AML) or mixed phenotype acute leukaemia (usually in the BM), and B- or T-lymphoblastic leukaemia/lymphoma (B-/T-ALL) in BM or in an extramedullary site. Thus it is critical to recognize the clinicopathologic features of these neoplasms, identify the molecular genetic lesions and classify them accordingly. An accurate diagnosis and classification have important therapeutic and prognostic implications.
Myeloid/Lymphoid Neoplasms Associated with PDGFRA Rearrangement
After imatinib was approved for chronic myeloid leukaemia (CML) use, it was tried in a number of disorders, including patients with hypereosinophilia. Some of the eosinophilic disorders were found to be extremely sensitive to imatinib, indicating the presence of an overactive tyrosine kinase that was inhibited [3, 4]. This led to the discovery of the chimeric oncoprotein encoded by FIP1L1-PDGFRA [5, 6]. It has been shown in human CD34+ cells that expression of FIP1L1-PDGFRA fusion protein induces cell proliferation and differentiation toward eosinophilic lineage in the absence of cytokines; these effects are mediated by the activation of NF-kB and STAT5 [7].
Epidemiology
PDGFRA rearranged cases overall are rare, but within the category of MLNE they are the most common. The most common fusion gene partner for PDGFRA is FIP1L1 [8]. It has been estimated that 10 to 20% of patients with unexplained eosinophilia in developed countries harbour PDGFRA rearrangements. There is a striking male predominance, with a male:female ratio of about 17:1. The median age at onset is in the late 40s, with most of patients between the ages of 25 and 55 years. Paediatric cases have also been reported [9]. Although the majority of cases are de novo, a subset of cases present as a therapy-related disease [10].
Clinical Features
In addition to eosinophilia that is detected in approximately 70% of patients [11], splenomegaly is found in about 60% of patients, followed by anaemia, thrombocytopaenia and neutrophilia. High level of vitamin B12 is characteristic. The disease at presentation may resemble chronic eosinophilic leukaemia (CEL, NOS) or idiopathic hypereosinophilic syndrome (HES), systemic mastocytosis (SM) with eosinophilia, CML, atypical chronic myeloid leukaemia, BCR-ABL1 negative (aCML), T-ALL, B-ALL, and rare cases may present as AML [12, 13], myeloid sarcoma [14] or concomitant AML and ALL [13]. One case of hypereosinophilia associated with a nodal angioimmunoblastic T-cell lymphoma has also been reported [15]. Patients presenting with AML or T-ALL often have significant eosinophilia (>1.5 × 109/L) before and/or at diagnosis of AML/T-ALL, or persistent eosinophilia after receiving intensive chemotherapy and achieving complete haematologic remission. These various case scenarios should trigger screening for PDGFRA rearrangements [13]. It should be kept in mind that cases of PDGFRA rearranged that are initially diagnosed as MPN, myelodysplastic/myeloproliferative neoplasm (MDS/MPN) or SM, may later progress to ALL or AML. Symptoms related to the release of eosinophilic granules commonly present as skin rash and erythema, and less frequently with pulmonary, gastrointestinal tract and/or cardiac manifestations. In untreated patients, or if patients have a long interval of disease before treatment, endomyocardial fibrosis and restrictive cardiomyopathy may occur.
Morphology
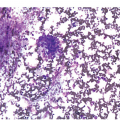
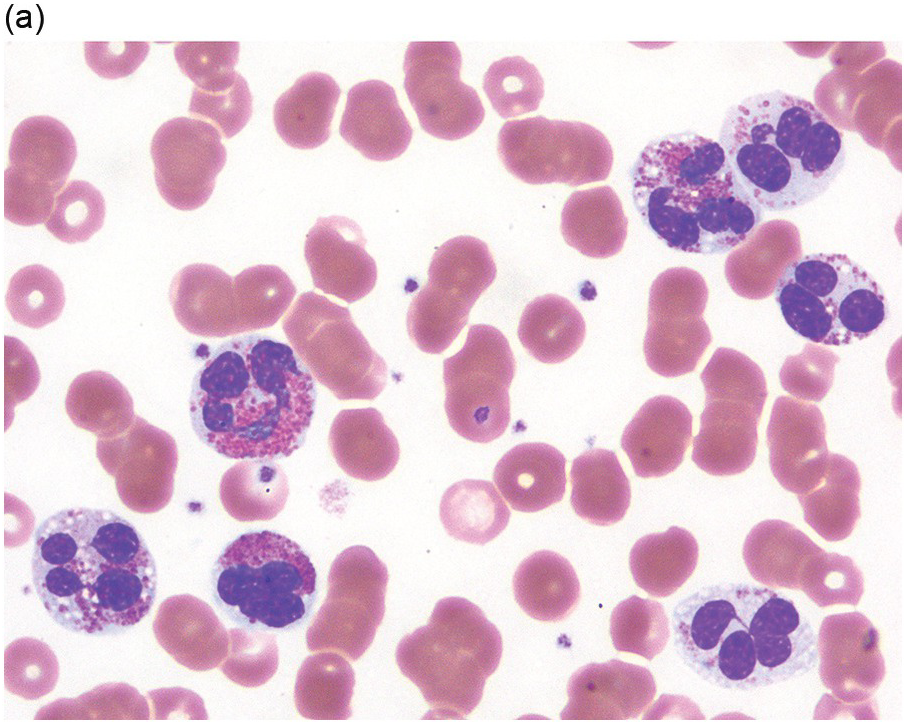
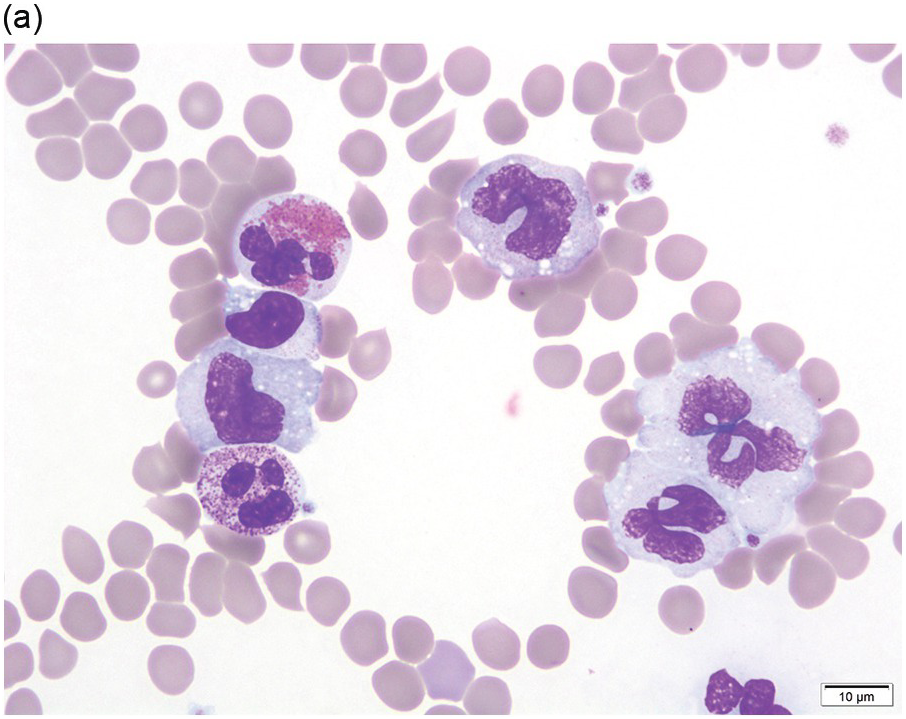
Peripheral blood shows eosinophilia with a predominance of mature forms; a variable number of eosinophilic myelocytes and promyelocytes can be seen. The eosinophils may exhibit a range of abnormalities including hypogranulation, degranulation, uneven granulation, cytoplasmic vacuolation, small or large granules, nuclear hyper- or hyposegmentation, and large or giant size (Figure 13.1). There is usually no basophilia.
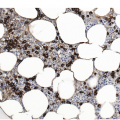
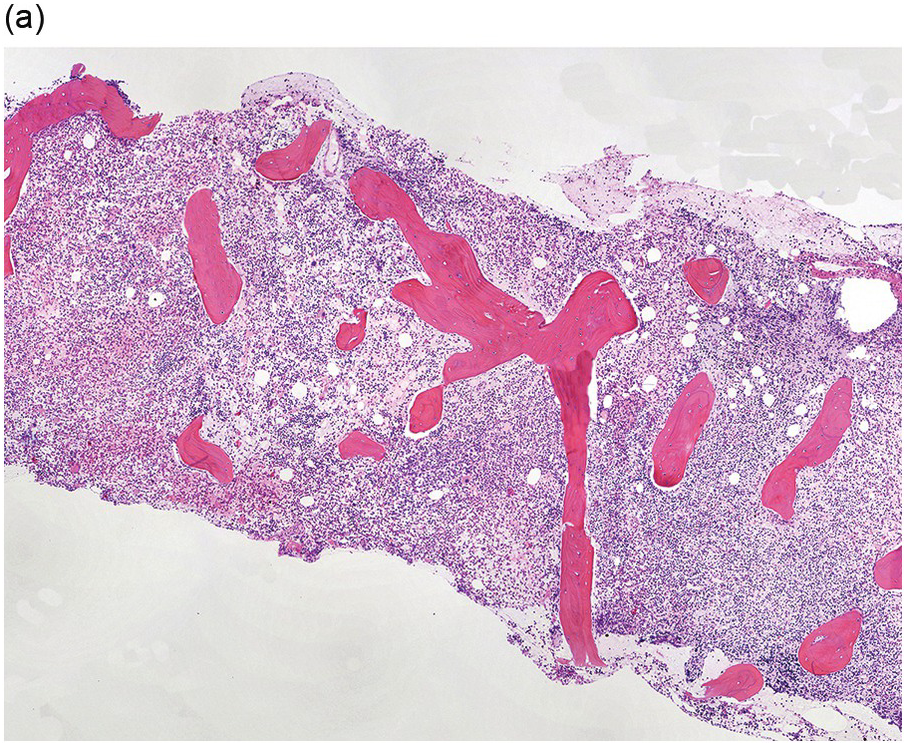
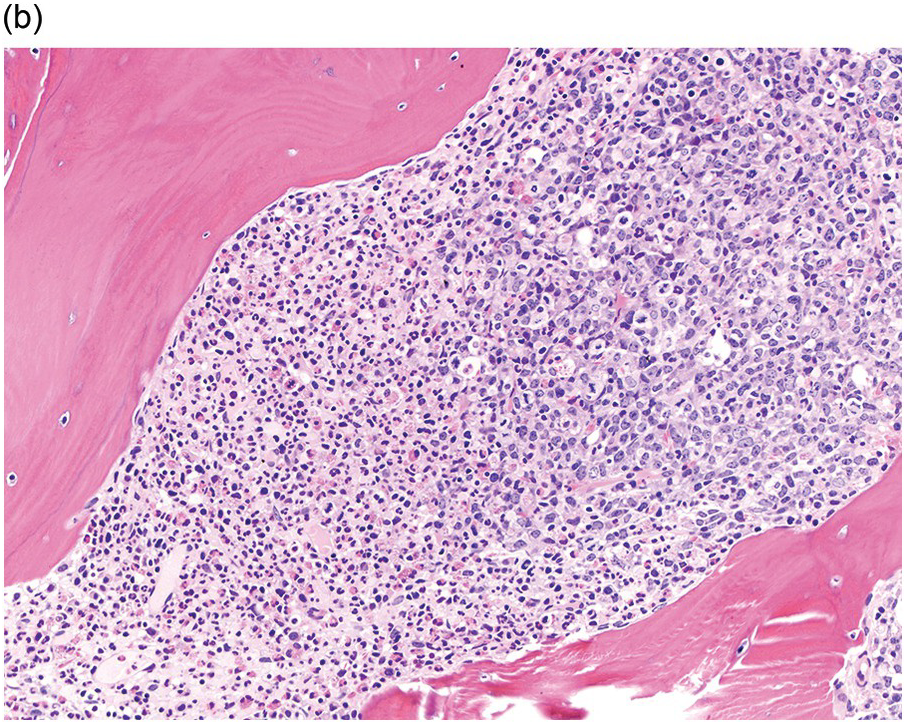
The BM is usually hypercellular with increased eosinophils and eosinophilic precursors (Figure 13.2a, b). Mature eosinophils may demonstrate abnormalities similar to those seen in the peripheral blood but often less pronounced. An excess of blasts is uncommon. Megakaryocytes can be unremarkable, but abnormalities may occur resembling megakaryocytes seen in MDS, MPN or MDS/MPN. Dysplastic features may include the presence of large megakaryocytes with separated nuclear lobes. Spindled mast cells are often observed but they are usually scattered and do not form compact aggregates (Figure 13.2c); immunophenotypically the mast cells show aberrant expression of CD25 in around 60% of cases. Marrow fibrosis of at least grade 1 of 3 is very common and is found in more than two-thirds of patients.
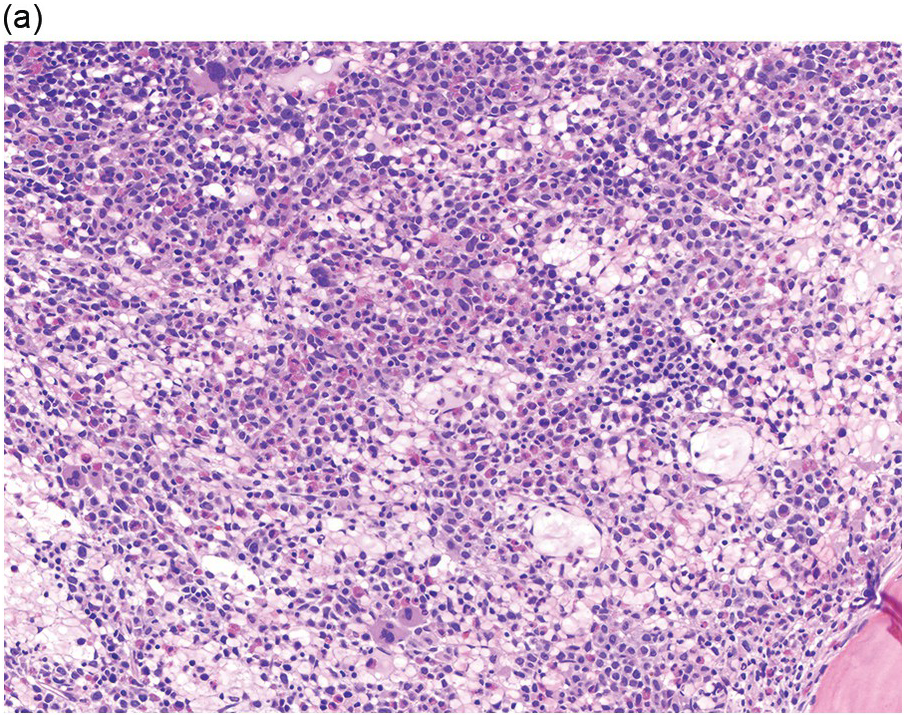
(a) Bone marrow shows hypercellularity. In this case megakaryocytes are unremarkable but sometimes they can be small hypolobated or large hypersegmented or with dysjoined lobes.
(b) Bone marrow shows eosinophilia and markedly decreased megakaryocytes. In other cases megakaryocytes can be increased, decreased or absent.

(c) Tryptase immunohistochemistry highlights scattered spindle mast cells; this is a common feature in this disease.
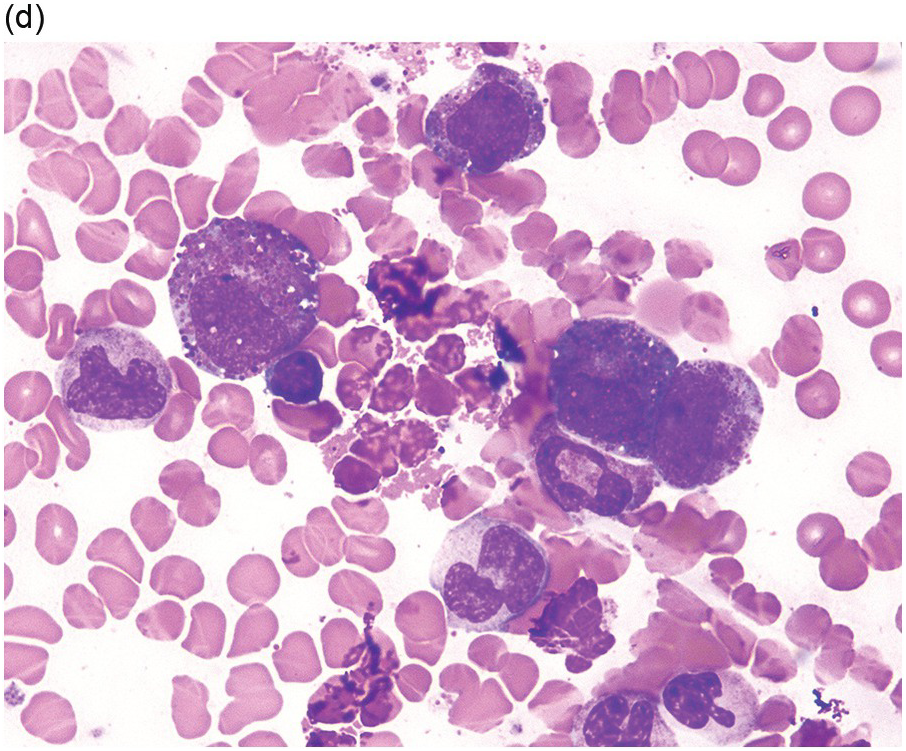
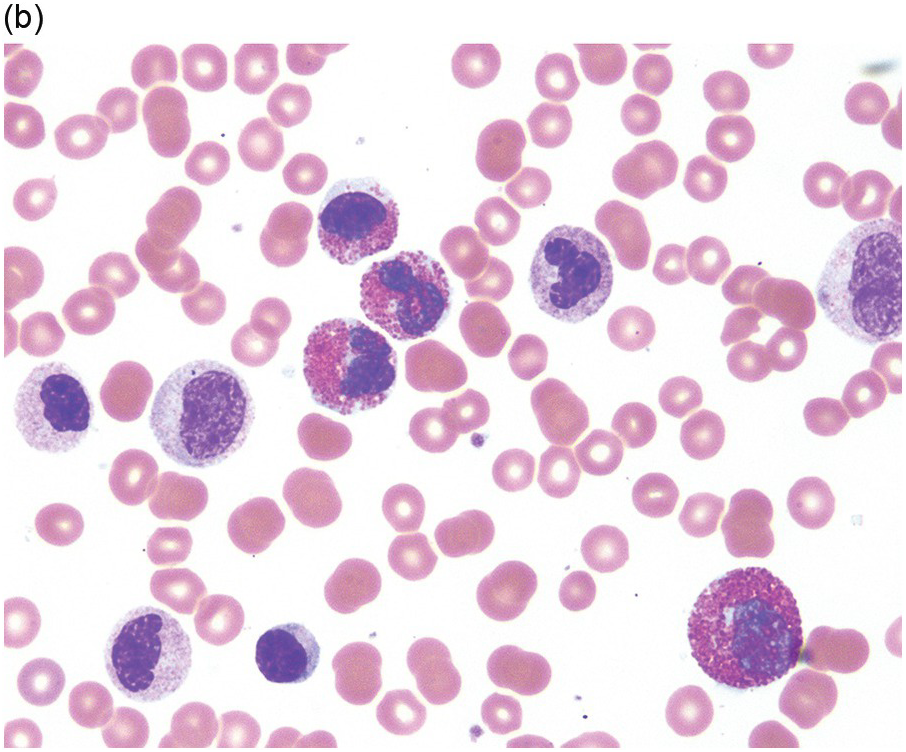
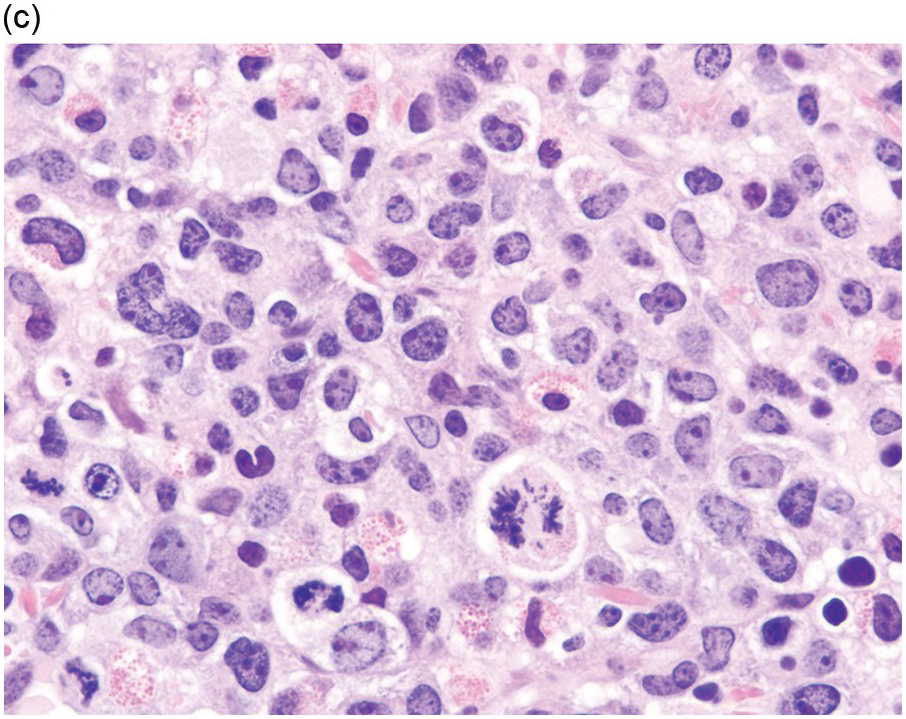
(d) Bone marrow aspirate smear shows increased eosinophils; precursor eosinophils may display abnormal basophilic granules, while granulocytes can show abnormal nuclear lobation.
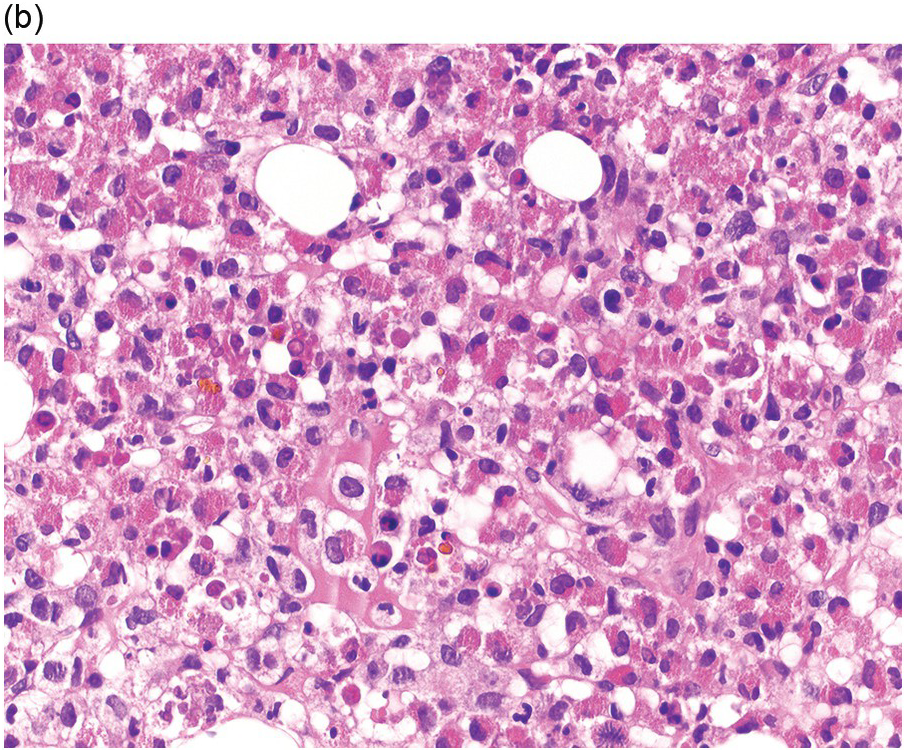
In addition to abnormal eosinophils, dysgranulopoiesis and dyserythropoiesis may be present. Granulocytes may show unusual nuclear segmentation and granulation (Figure 13.2d). Blasts may be increased. Some cases present or transform in extramedullary sites such as the epidural space, which can lead to a diagnosis of myeloid sarcoma (Figure 13.3).
Immunophenotype
Eosinophils are markedly increased in BM specimens. Compared to neutrophils, eosinophils have high side scatter, bright CD45 and co-expression of CD4 and, in contrast with neutrophils, eosinophils are negative for CD10, CD15, CD16 and CD64 (Figure 13.4a). However, these markers do not differentiate neoplastic eosinophils from reactive/secondary eosinophils. The CD34+ myeloblasts are often not increased in the chronic phase of the disease but often demonstrate an abnormal immunophenotype (Figure 13.4b). The presence of scattered spindle mast cells with aberrant CD25 and/or CD2 expression should raise the suspicion of an associated myeloid neoplasm. Immunophenotypic studies for T-cells and mast cells are encouraged. The former is to exclude the possibility of lymphocytic-variant hypereosinophilic syndrome or hypereosinophilia (HE) associated with a T-cell neoplasm.
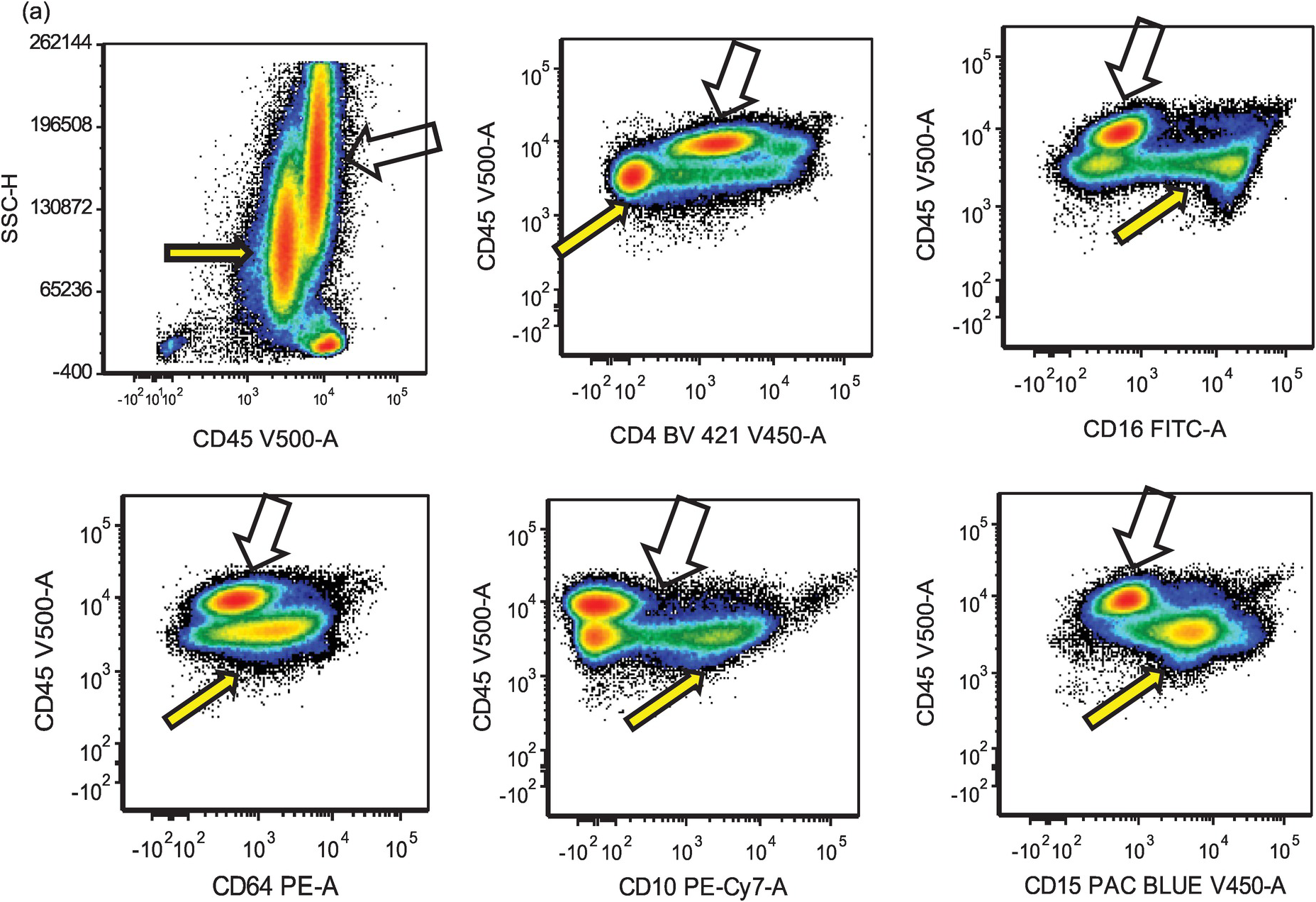
Figure 13.4(a) MLNE and PDGFRA rearrangement: flow cytometric features. The immunophenotype of eosinophils (empty arrows) is bright CD45+, CD4+, CD16–, CD64–, CD10– and CD15–; in comparison, neutrophils and precursors (yellow arrows) show dim CD45, CD4–, CD16+, CD64+, CD10+ and CD15+. However, these markers do not differentiate neoplastic eosinophils from normal/reactive eosinophils.
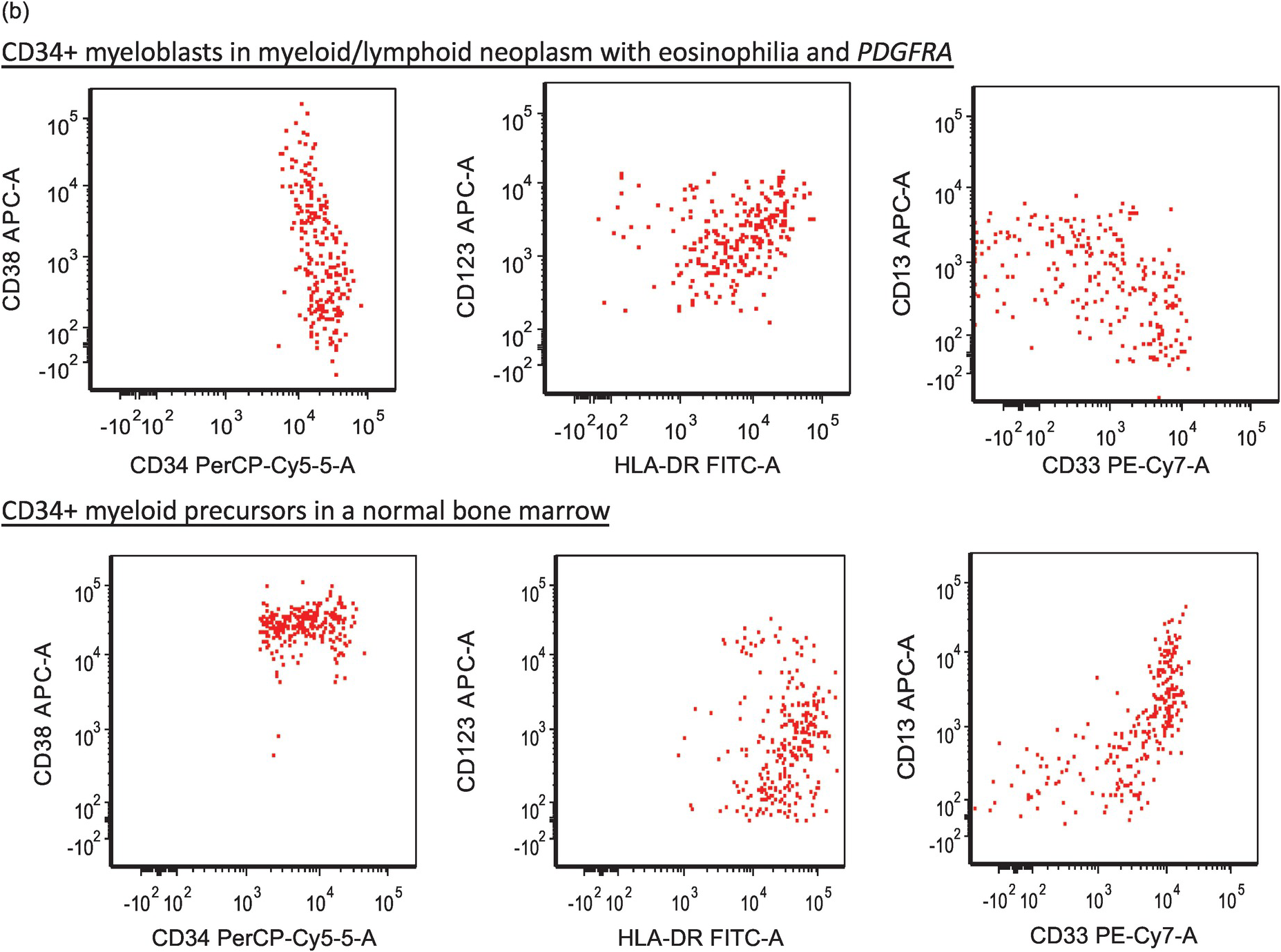
Figure 13.4(b) Immunophenotypic aberrancies of myeloblasts in cases of MLNE and PDGFRA rearrangement. The upper panel displays aberrant blasts in a case with PDGFRA rearrangement. It shows that the total number of CD34+ myeloblasts is not increased (total, 0.2%); however, these blasts are aberrant in showing decreased CD38, increased CD34, CD123, decreased HLA-DR expression as well as a disrupted CD13/CD33 pattern. The lower panel is a normal BM in which myeloid precursors exhibit normal expressions of CD38, CD34 and CD123, and a normal CD13/CD33 pattern.
Cytogenetics and Molecular Genetics
The interstitial deletion of 800 kb that leads to FIP1L1–PDGFRA fusion is undetectable by conventional cytogenetics because the level of resolution of conventional cytogenetics is in the range of 10 megabases. Therefore conventional cytogenetics often shows a diploid karyotype or unrelated clonal abnormality such as trisomy 8 in cases with FIP1L1–PDGFRA. Routine detection of the interstitial deletion in clinical practice is best achieved by interphase or metaphase fluorescence in situ hybridization (FISH). Since the CHIC2 gene is contained in the deleted region, the FISH test to detect FIP1L1–PDGFRA gene fusion is often referred to as ‘CHIC2 deletion’ (Figure 13.5). The fusion also can be detected by reverse transcriptase-polymerase chain reaction (RT-PCR) [5]. The fusion can be detected in peripheral blood, BM specimens or in involved tissues.
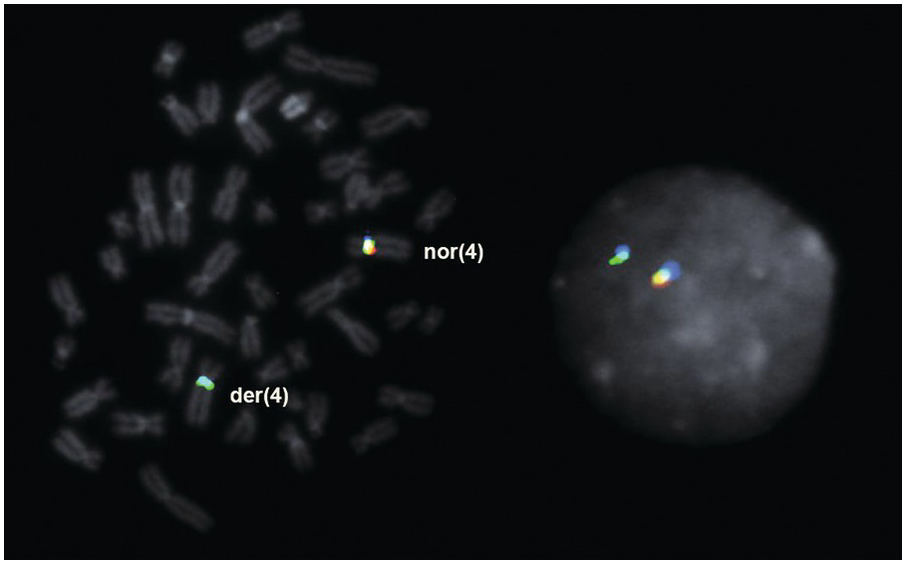
Figure 13.5 MLNE with PDGFRA rearrangement: FISH findings. Karyotype is diploid; the 0.8-megabase microdeletion was below the level of detection of 10 megabases for conventional cytogenetics. FISH using a three-colour probe for FIP1L1 (green), CHIC2 (red) and PDGFRA (aqua) demonstrates loss of CHIC2 and fusion of FIP1L1 and PDGFRA, consistent with FIP1L1-PDGFRA.
Other than FIP1L1, other partner rearrangements for PDGFRA have been reported (Table 13.1) [16–20]. These include KIF5B-PDGFRA [16], CDK5RAP2-PDGFRA associated with ins(9;4)(q33;q12q25) [17], STRN-PDGFRA fusion as a result of t(4;12)(q12;p13.2) [21];TNKS2-PDGFRA associated with t(4;10)(q12;q23.3) [18] and ETV6-PDGFRA [22]. The cases with these variant rearrangements often have a clinical and morphological presentation of CEL, NOS [23]. BCR-PDGFRA resulting from t(4;22)(q12;q11.2)[6, 24–26] may have a clinical presentation of atypical CML with monocytosis, or hybrid features of CEL and CML, or T-lymphoblastic leukaemia/lymphoma. Cases with variant rearrangements are also sensitive to imatinib therapy [5, 10, 16].
Table 13.1 Gene partners of myeloid and lymphoid neoplasms with PDGFRA rearrangements.
| Reference | Chromosomal abnormality | Gene partner | Clinical features | Response to TKI |
|---|---|---|---|---|
| Cools et al. [5] | 4q12 deletion; rare t(1;4) (q44;q12) | FIP1L1 | CEL; most frequent variant | Yes, imatinib; resistant with T674I mutation |
| Curtis et al. [21] | t(2;4)(p24;q12) | STRN | CEL | Yes, imatinib |
| Sugimoto et al. [20] | t(3;4)(p13;q12) | FOXP1 | CEL | Yes, imatinib |
| Score et al. [16] | t(4;10)(q12;p11) | KIF5B | CEL | Yes, imatinib |
| Chalmers et al. [18] | t(4;10)(q12;q23.3) | TNKS2 | CEL; KRAS G12D mutation | Yes, imatinib |
| Curtis et al. [21] | t(4;12)(q12;p13.2) | ETV6 | CEL | Yes, imatinib |
| Zhou et al. [23] | t(4;14)(q12;q24) | Not determined | Therapy-related AML with eosinophilia | Not tested |
| Safley et al. [10] | t(4;22)(q12;q11.2) | BCR | Leukocytosis with monocytosis; DLBCL therapy related | Yes, imatinib |
| Walz et al. [17] | ins(9;4)(q33;q12q25) | CDK5RAP2 | CEL | Yes, imatinib chronic phase; resistant on blast phase |
CEL: chronic eosinophilic leukaemia; aCML: atypical chronic myeloid leukaemia
DLBCL: diffuse large B-cell lymphoma
TKI: tyrosine kinase inhibitor
Any haematological malignancy, if in doubt, should be screened for the cytogenetically cryptic FIP1L1-PDGFRA fusion gene by RT-PCR and/or FISH analysis, as affected patients are excellent candidates for treatment with tyrosine kinase inhibitors.
Prognosis
Almost all patients with the FIP1L1-PDGFRA fusion are sensitive to tyrosine kinase inhibitors such as imatinib [5, 27, 28]. Primary or secondary resistance to imatinib is unusual in these patients [5, 29, 30]. Discontinuation of therapy can lead to relapse of disease in a subset of patients [31].
Resistance involves the T674I mutation within the ATP-binding domain of PDGFRA, which is analogous to the T315I in ABL1 mutation in CML and confers resistance to the TKIs [5, 29]. Additional chemotherapy may be needed when patients present as acute leukaemia.
Myeloid and Lymphoid Neoplasms Associated with PDGFRB Rearrangement
PDGFRB is located at chromosome 5q32, and the breakpoints range between 5q31 and 5q33. The most common translocation is t(5;12)(q32;p13.2), resulting in ETV6-PDGFRB gene fusion. Due to frequent presentations of monocytosis and eosinophilia, cases with t(5;12)(q32;p13.2) were in the past classified as chronic myelomonocytic leukaemia (CMML) with eosinophilia associated with t(5;12). More than 30 other partner genes have been found fused with PDGFRB, and considered as uncommon variants (Table 13.2) [32–66]. However, cases of B-ALL with an immunophenotype or gene signature of BCR-ABL1-like are specifically excluded from this category; these include EBF1-PDGFRB, SSBP2-PDGFRB, TNIP1-PDGFRB, ZEB2-PDGFRB and ATF7IP-PDGFRB [10, 67, 68, 69].
Table 13.2 Gene partners of myeloid and lymphoid neoplasms with eosinophilia and PDGFRB rearrangement.
| Reference | Chromosomal rearrangement | Gene partner | Clinical features | Response to TKI |
|---|---|---|---|---|
| Hidalgo-Curtis et al. [37] | t(1;3;5)(p36;p22.2;q32) | WDR48 | CEL, AML | Yes, imatinib |
| Walz et al. [39] | der(1 )t(1 ;5)(p34;q33), der(5)t(1;5)(p34;q15), der(11)ins(11;5)(p13;q15q32) | CAPRIN1 (GPIAP1) | CEL | Yes, imatinib |
| Saultz et al. [40] | Inv(5)(q13q33) | Unknown | CEL | Yes, imatinib |
| Rosati et al. [41] | t(1;5)(q21.3;q32) | TPM3 | CEL | Yes, imatinib |
| Wilkinson et al. [42] | t(1;5)(q23;q33) | PDE4DIP | MDS/MPN with eosinophilia | Yes, imatinib |
| Gallagher et al. [43] | t(2;5)(p16.2;q32) | SPTBN1 | CEL | Yes, imatinib |
| Naumann et al. [32] | t(3;5)(q13;q33) | GOLGB1 | MPN with eosinophilia | Yes, imatinib |
| Walz et al. [39] | t(4;5;5)(q21.2;q31 ;q32) | PRKG2 | Chronic basophilic leukaemia | Yes, imatinib |
| Gallagher et al. [43] | t(4;5)(q21;q33) | PRKG2 | CEL | Yes, imatinib |
| Campregher et al. [44] | t(3;5)(q26;q32) | TBL1XR1 | MPN with eosinophilia post AML with t(6;9) | Yes, dasatinib |
| Hidalgo-Curtis et al. [37] | t(3;5)(p21;q21) | GOLGA4 | CEL or aCML with eosinophilia | Yes, imatinib |
| Ross et al. [45] | Cryptic interstitial deletion of 5q | TNIP1 | CEL with thrombocytosis | Yes, imatinib |
| Winkelmann et al. [46] | t(5;6)(q33;q22) | CEP85L | CEL | Yes, imatinib |
| Ross et al. [47] | t(5;7)(q33;q11.2) | HIP1 | CMML with eosinophilia | NA |
| Medves et al. [48] | t(5;9)(q31–32;p22–24) | KANK1 | Thrombocythaemia; no eosinophilia | Yes, imatinib |
| Schwaller et al. [49] | t(5;10)(q32;q21.2) | CCDC6/H4 | MPN with eosinophilia | NA |
| Erben et al. [50] | 46,XY[4] | SART3 | MPN with eosinophilia and myelofibrosis | Yes, imatinib |
| Zou et al. [36] | t(5;11)(q32;q13.4) | NUMA1 | MPN with response to eosinophilia | |
| Hidalgo-Curtis et al. [37] | t(5;12)(q32;q13) | BIN2 | aCML with eosinophilia, skin rash | Yes, imatinib |
| Bidet et al. [51] | t(5;12)(q33;p13) | ETV6 | CMML with eosinophilia; most common in this category | Yes |
| Naumann et al. [32] | t(5;17)(q33;p15) | GPSF6 | MPN with eosinophilia | Yes, imatinib |
| Walz et al. [39] | t(5;12)(q31–33;q24) | GIT2 | CEL | Yes, imatinib |
| Gorello et al. [65] | t(5;12)(q33;p13.3) | ERC1 | AML; no eosinophilia | Yes, imatinib |
| Vizmanos et al. [52] | t(5;14)(q32;q22.1) | NIN | MPN with eosinophilia | Yes, imatinib |
| Levine et al. [53] | t(5;14)(q33;q32) | CCDC88C (KIAA1509) | CMML with eosinophilia | Yes, imatinib |
| Kim et al. [34] Gong et al. [66] | t(5;14)(q33;q32) | TRIP11 (CEV14) | MPN Therapy-related APL | Yes, imatinib |
| Grand et al. [54] | t(5;15)(q33;q22) | TP53BP1 | CEL | Yes, imatinib and subsequent resistance |
| La Starza et al. [55] | t(5;16)(q33;p13) | NDE1 | MPN with eosinophilia | Yes, imatinib |
| Naumann et al. [32] | t(5;17)(q33;p11) | MPRIP | MPN with eosinophilia | Yes, imatinib |
| Magnusson et al. [56] | t(5;17)(q32;p13.2) | RABEP1 | CMML; no eosinophilia | Yes, imatinib |
| Morerio et al. [57] | t(5;17)(q33;p11.2) | SPECC1 (HCMOGT-1) | JMML with eosinophilia | Not tested |
| Sheng et al. [58] | t(5;17)(q32;q11.2) | MY018A | MPN with eosinophilia | Yes, imatinib |
| Greco et al. [59] | t(5;17)(q32;q21.3) | COL1A1 | MDS or MPN with eosinophilia | |
| Patnaik et al. [60] | t(5;19)(q33;13.3) | Unknown | CMML; no eosinophilia | Yes, imatinib |
| Gosenca et al. [33] | t(5;20)(q32;p11.2) | DTD1 | CMML | Yes, imatinib |
| Bell et al. [61] | Del(7)(q22q32) | SDPR | CMML no eosinophilia, cryptic rearrangement | Partial response to imatinib |
| Cases with PDGFRB rearrangements and BCR-ABL1-like B-ALL phenotypea | ||||
| Schwab et al. [62]; Weston et al. [63] | EBF1-PDGFRB | B-ALL | Response to imatinib | |
| Zhang et al. [64] | ATF7IP- PDGFRB | B-ALL | Response to imatinib | |
a These cases present with BCR-ABL1-like B-ALL phenotype and should not be classified in the category of lymphoid/myeloid neoplasms with PDGFRB rearrangement
AML: acute myeloid leukaemia; CEL: chronic eosinophilic leukaemia; aCML: atypical chronic myeloid leukaemia
CMML: chronic myelomonocytic leukaemia; JMML: juvenile myelomonocytic leukaemia; MPN: myeloproliferative neoplasm
Epidemiology
PDGFRB rearrangements associated with 5q31–33 translocations are rare, with a reported incidence of 1.8% of all MPNs [70, 71], and are identified in less than 3% of patients with unexplained eosinophilia and <1% of patients with CMML; more rare cases occur secondary to therapy for AML [44, 72]. Most affected patients are adult men, with a median age in the fifth decade of life [70, 73, 74].
Clinical Features
Patients usually present with anaemia, leukocytosis, monocytosis, eosinophilia and splenomegaly [70, 75]. Eosinophilia is common, but not invariably present in cases with PDGFRB rearrangement, and it has been estimated to occur in 60% of cases [ 76]. These neoplasms at the time of presentation most often resemble CMML, CEL-NOS, BCR-ABL1 negative atypical CML, or occasionally MDS. Presentation as AML is uncommon. T-ALL has been occasionally reported at presentation or at progression, but cases of PDGFRB rearrangement presenting as B-ALL usually fall in the category of BCR-ABL1-like B-ALL [69].
Similar to the cases of rearranged PDGFRA, if patients have a long duration of hypereosinophilia prior to treatment, symptoms and tissue damage due to eosinophilic infiltration, activation and degranulation often occur. Skin lesions are the most common, cardiac injury may be observed. Splenomegaly is common and hepatomegaly may be present.
Morphology
A significant proportion of patients present as CMML with eosinophilia or hypereosinophilia with monocytosis. In peripheral blood, both eosinophils and monocytes may exhibit abnormal features (Figure 13.6). Some cases would be classified as CEL or some form of MPN with increased eosinophils if the diagnostician is blinded to molecular genetic information. Eosinophil abnormalities include abnormal granulation, nuclear lobation, large size and left-shifted maturation. Some patients may show a high WBC count with marked dysgranulopoiesis and left-shifted myeloid maturation, with haematological features of atypical CML with eosinophilia. These morphological entities often show eosinophils in peripheral blood and/or BM; however, eosinophilia may not necessarily to be so pronounced in every case (≥1.5 × 109/L).
(a) Peripheral blood shows leukocytosis and monocytosis. Monocytes exhibit abnormal nuclear lobation.
(b) Bone marrow biopsy showing hypercellularity. Megakaryocytes are large with separated nuclear lobes.
(c) Bone marrow aspirate smear shows monocytosis and eosinophilia. Monocytes are mature but show abnormal nuclear shapes.
(d) Immunohistochemistry for tryptase demonstrates scattered spindle-shaped mast cells in a loose cluster.
Spindle mast cells may be observed, similar to PDGFRA rearranged cases. In some cases, mast cells may form large clusters, spindle with aberrant CD25 expression and fulfil the diagnostic criteria of systemic mastocytosis associated with hypereosinophilia.
Immunophenotype
Similar to cases with rearranged PDGFRA, immunophenotypic studies for T-cells and mast cells are encouraged. The former is to exclude the possibility of lymphocytic-variant hypereosinophilic syndrome or hypereosinophilia (HE) associated with a T-cell neoplasm. A small number of mast cells with an aberrant expression of CD25 may be detected, which provides a hint for a myeloid neoplasm that requires further molecular genetic characterization. If blasts are increased, a comprehensive immunophenotypical analysis should be performed, aiming to characterize the blast immunophenotype. If blasts have an immunophenotype consistent with B-ALL, the case may be better classified as BCR-ABL1-like B-lymphoblastic leukaemia, and a search made for one of the following fusions: EBF1-PDGFRB, SSBP2-PDGFRB, TNIP1-PDGFRB, ZEB2-PDGFRB and ATF7IP-PDGFRB [67, 68, 69].
Cytogenetics and Molecular Genetics
The most common translocation involving PDGFRB is t(5;12)(q33;p13)/ETV6-PDGFRB. It is believed that conventional cytogenetic analysis reveals 5q31–33 abnormalities in all cases if sufficient metaphases are obtained for analysis. However, by RNA sequencing, PDGFRB rearrangement has been detected in cases with no apparent karyotypic abnormality involving 5q31–33 [77], suggesting a genome-wide or targeted RNAseq may be the best option for the detection of these TK fusions. On the other hand, not all translocations characterized as t(5;12)(q31–33;p12) lead to ETV6-PDGFRB fusion. Therefore FISH study is recommended to confirm that PDGFRB is rearranged in cases with breaks at 5q31–33. The breakpoints can be detected using FISH with break-apart probes for the PDGFRB locus; however, this approach does not identify the gene partner. More than 30 different gene partners of PDGFRB have been identified (Table 13.2). These gene fusions lead to the creation of chimeric proteins that have enhanced tyrosine kinase activity. The caveat is that FISH analysis is not 100% sensitive to demonstrate rearrangement of PDGFRB [74]. RT-PCR analysis using primers covering all possible breakpoints of ETV6-PDGFRB is recommended for suspicious cases or for confirmation; alternatively for cases in which this specific translocation cannot be confirmed, a therapeutic trial with imatinib is recommended, for practical purposes [22].
It is of interest that Galimberti et al. [78] reported that 9 of 29 (31.0%) patients with myelodysplastic syndromes and 2 of 7 (28.6%) patients with CMML, all with karyotypes not involving 5q31–33, and none with eosinophilia, had rearrangements of PDGFRB as determined by FISH; the median percentage of affected cells was 5% (range, 3–9%). Surprisingly, a subset of cases with the rearrangement had improvement of cytopaenias or transfusion dependence, but it was acknowledged that the data was very preliminary.
Prognosis
Most haematolymphoid neoplasms associated with translocations of PDGFRB are sensitive to tyrosine kinase inhibitors. Patients with these neoplasms respond to imatinib therapy with excellent haematologic, cytogenetic and molecular responses [76]. Long-term follow-up of patients with rearranged PDGFRB patients treated with imatinib reported a 96% response rate and a ten-year overall survival rate of 90% [79]. Primary or secondary resistance to imatinib is uncommon [73, 80]. When patients present with blast phase or develop acute leukaemia, chemotherapy in addition to tyrosine kinase inhibitors is needed [44].
Myeloid/Lymphoid Neoplasms Associated with FGFR1 Rearrangement
An association between the t(8;13) and the triad of T-lymphoblastic lymphoma/leukaemia, eosinophilia and myeloid malignancy was first reported in 1992 [81]. MLNE and FGFR1 rearrangement are derived from pluripotent haematopoietic stem cells and cases can present as an MPN or AML, T- or B-ALL/L or mixed-lineage acute leukaemia associated with peripheral blood and/or BM eosinophilia [1]. The latter may be an indication for the blast phase of an undiagnosed/underlying MPN. These features led to its former designation as stem cell myeloproliferative disorder or 8p11 myeloproliferative syndrome [82]. The diagnostic criteria for MLNE and FGFR1 rearrangement has been proposed in the 2016 revised WHO classification of haematolymphoid neoplasms [1]. (1) A myeloproliferative (MPN) or myelodysplastic/myeloproliferative neoplasm (MDS/MPN) with prominent eosinophilia and sometimes with neutrophilia or monocytosis, or (2) AML, T- or B-ALL/L, or mixed-phenotype acute leukaemia (usually associated with peripheral blood or BM eosinophilia); and (3) the presence of t(8;13)(p11.2;q12) or a variant translocation leading to FGFR1 rearrangement, demonstrated in myeloid cells, lymphoblasts or both.
Epidemiology
Myeloid/lymphoid neoplasms associated with FGFR1 rearrangement are rare. This disease affects patients with a wide range of ages (3–84 years, median 30–40 years) with a slight male predominance (1.5:1) [82, 83].
Clinical Features
Patients usually present with B-type symptoms [84] including fatigue, night sweats, weight loss and fever. Lymphadenopathy and hepatosplenomegaly are common, especially in patients with a T-lymphoblastic lymphoma presentation.
Morphology
Depending on the presentation, myeloid/lymphoid neoplasms associated with FGFR1 rearrangement may show different morphological findings in peripheral blood, BM or extramedullary sites [85]. Patients with a MPN presentation usually show peripheral eosinophilia, neutrophilia and occasionally monocytosis. In such cases the BM is usually hypercellular with eosinophilia and other features of MPN, such as increased megakaryocytes with clustering, increased myeloid, stromal cells and vascularity, and fibrosis. Patients who presented with chronic phase of disease may progress to AML or lymphoblastic leukaemia/lymphoma after a short interval.
In patients with an acute leukaemic presentation, the blasts may be B-lymphoblasts, T-lymphoblasts, mixed phenotype/mixed lineage leukaemia or AML. The background features of MPN may be obscured by the presence of large numbers of blasts or considered as changes associated with acute leukaemia. After obtaining morphological remission post high-dose chemotherapy, the peripheral blood and BM features of MPN often become more obvious; if not immediately, the changes will ensue over time. Increased eosinophils are frequently present; however, their level may not reach 1.5 × 109/L.
Myeloid/lymphoid neoplasms associated with FGFR1 rearrangement frequently present with nodal or extranodal disease (Figure 13.7), especially those associated with t(8;13)(p11;q12)/ZMYM1-FGFR1. Most nodal or extranodal manifestations have a predominant T-lymphoblastic component admixed with scattered or perivascular myeloid blasts. Because of the separate myeloid and T-cell components identified in lymph nodes, the term of bilineal lymphoma has been used [86–88]. The presence of the immature myeloid component may lead to a diagnosis of myeloid sarcoma. In these nodal and extranodal diseases, an eosinophilic infiltrate is frequently present that may provide a hint for the diagnosis. In these patients, peripheral blood and BM may show features of a CML, CEL, MPN-unclassifiable, aCML or MDS/MPNs.
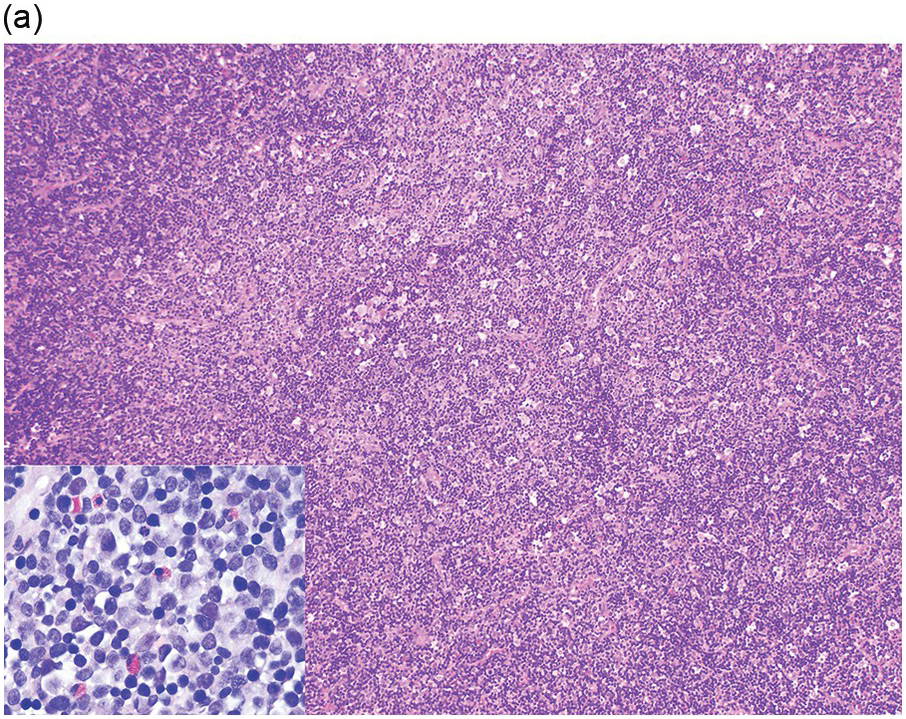
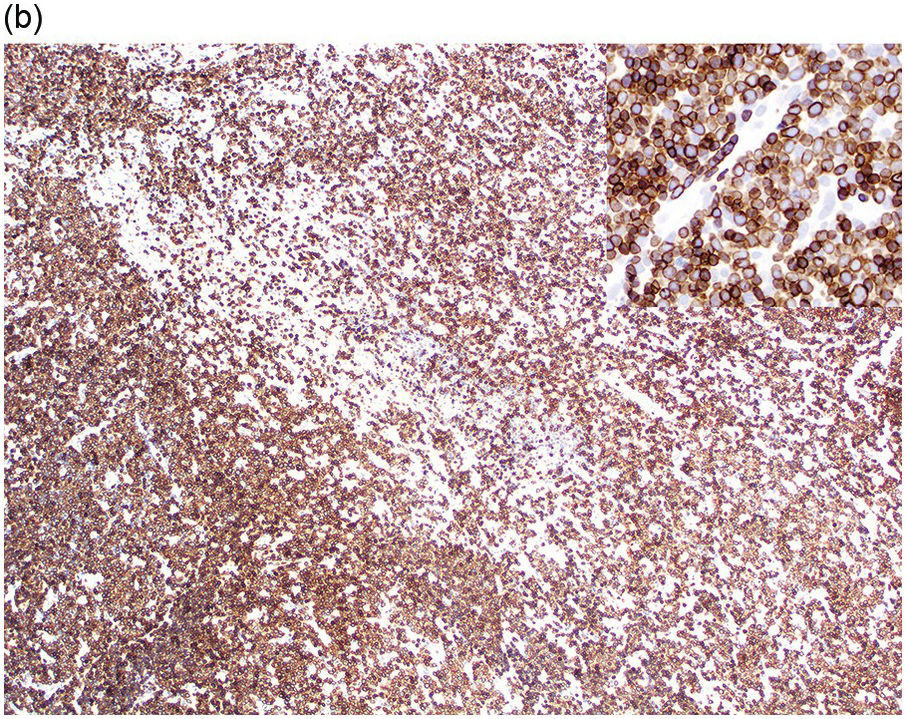
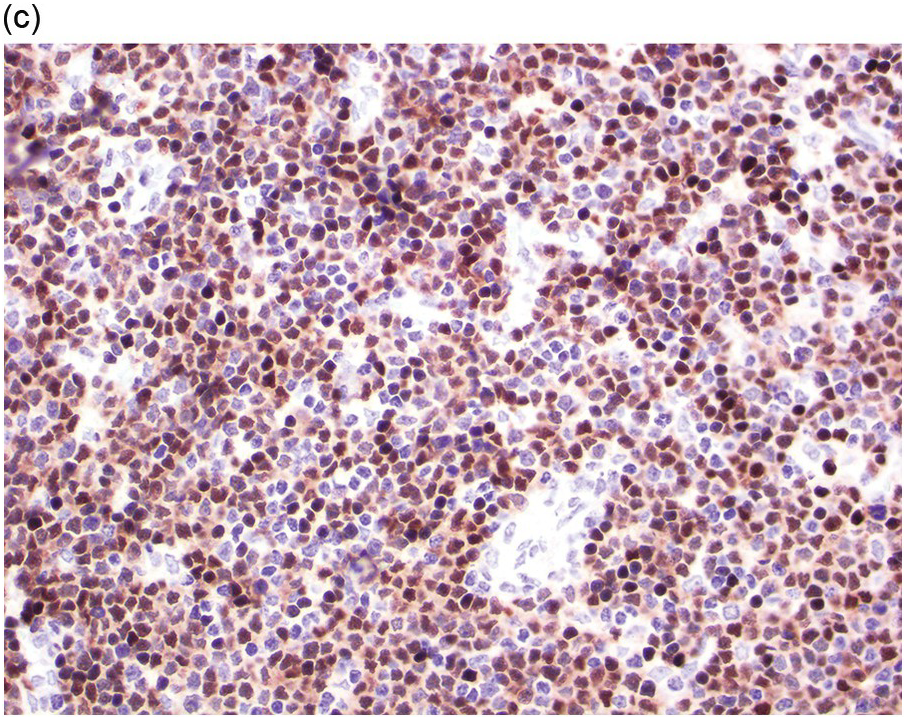
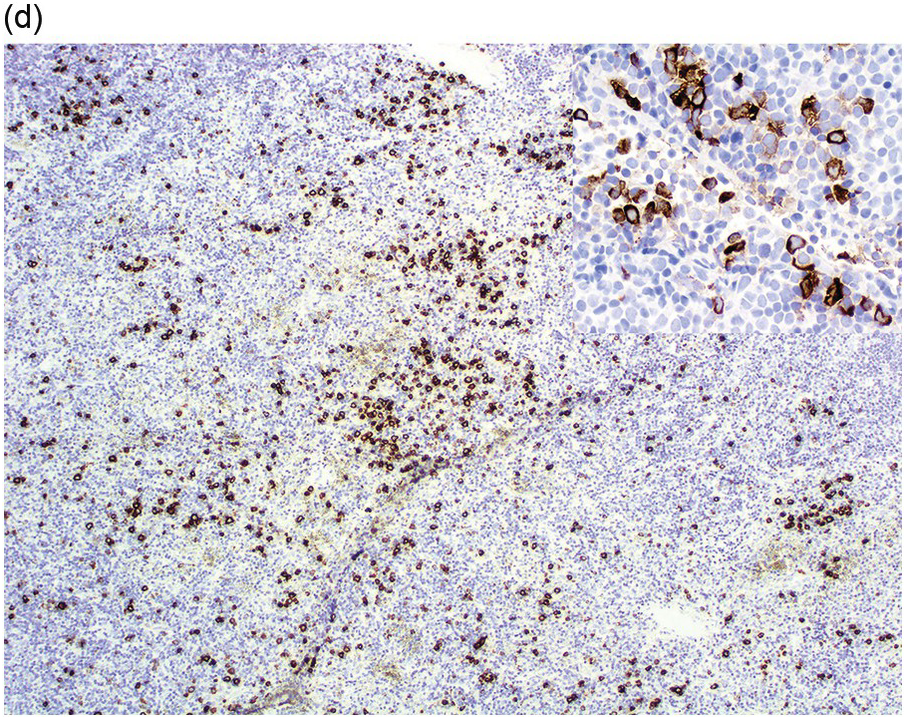
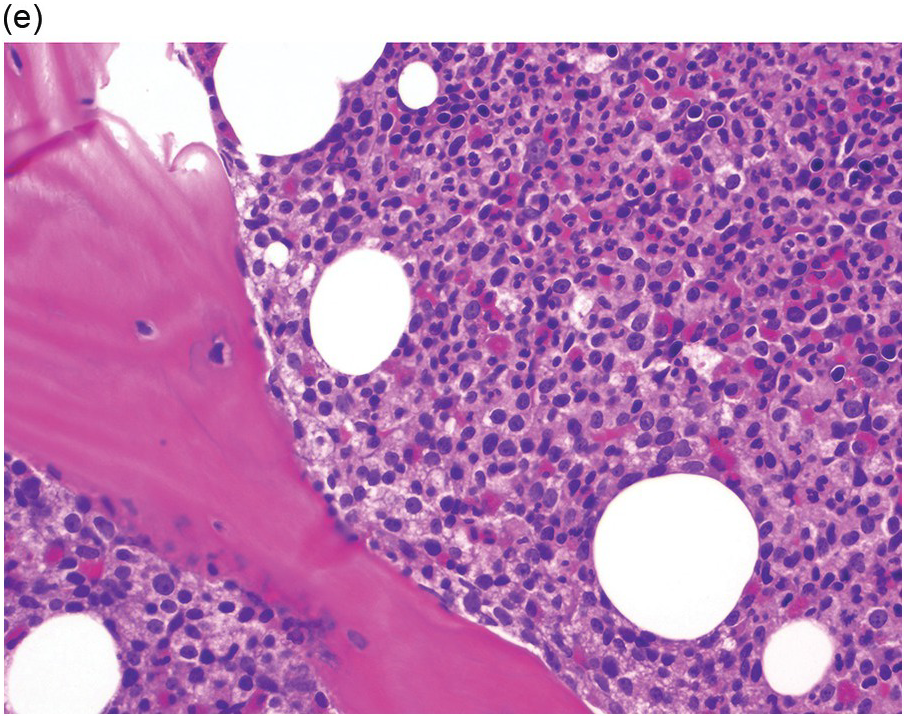
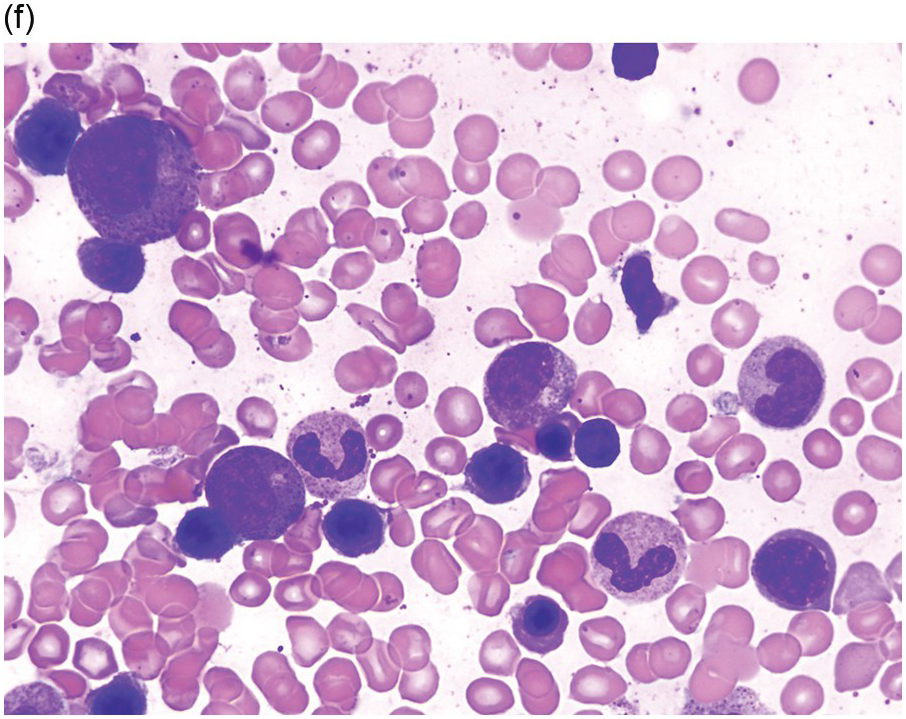
Figure 13.7 MLNE with FGFR1 rearrangement, with a lymphoid and myeloid blast phase of disease. (a) Lymph node is diffusely infiltrated by immature mononuclear cells. Higher magnification shows numerous small blasts and scattered eosinophils (inset). (b & inset) Immunohistochemistry for the T-cell marker CD3 highlights most cells in the infiltrate. (c) Immunohistochemistry for the immature lymphoid-cell marker TdT highlights most cells in the infiltrate and supports they are lymphoblasts. (d & inset) Immunohistochemistry for myeloperoxidase highlights scattered immature mononuclear cells, supporting myeloid lineage involvement. (e) Bone marrow biopsy shows hypercellularity with increased neutrophils and eosinophilia, consistent with a myeloproliferative neoplasm. (f) Bone marrow aspirate smear shows eosinophil precursors with basophilic granules.
Immunophenotype
As a component of the immunophenotypic analysis of T-cells and mast cells that is recommended for all patients with hypereosinophilia, the blasts need also to be analyzed for immunophenotypic aberrancies that may disclose evidence of another underlying neoplasm, even when blasts are not increased. The blasts may display a typical immunophenotype of T-lymphoblasts, B-lymphoblasts or myeloblasts. For cases that present as ALL, they may have a very minor population of aberrant myeloblasts, similar to CML with lymphoid blast phase. In the lymph node, a detailed immunophenotype often will show that both types of blasts are admixed.
Cytogenetics and Molecular Genetics
Conventional cytogenetic analysis is the most reliable technique to demonstrate translocations at 8p11 in the majority of cases with rearrangements of FGFR1. The t(8;13) is the most common translocation observed in this disease, in which ZMYM2 is the most common partner of FGFR1. Patients with the ZMYM2-FGFR1 rearrangement most often present with an MPN with eosinophilia and concurrent T-lymphoblastic lymphoma; rapid evolution to blastic T/myeloid neoplasm is also common, for which the name of blastic T/myeloid neoplasm with ZMYM2-FGFR1 has been suggested [85].
The diagnosis of myeloid/lymphoid neoplasm with FGFR1 rearrangement can be confirmed by using FISH with break-apart probes for FGFR1. There are 14 partners that can fuse with the FGFR1 (Table 13.3) [81, 89–101]. However, clinicopathological manifestations may differ according to the gene partners. For example, patients with myeloid/lymphoid neoplasms with t(8;22)(p11.2;q11.2)/BCR–FGFR1, usually manifest with monocytosis and the blast phase is predominantly B-ALL [100, 102]. Patients with t(6;8)(q27;p11.2) may present with polycythaemia [103].
Table 13.3 Gene partners of FGFR1 in myeloid and lymphoid neoplasms with eosinophilia.
| Reference | Chromosomal abnormality | Gene partner | Name of gene partner | Clinical features | Response to TKI |
|---|---|---|---|---|---|
| Kim et al. [89] | t(1;8)(q25;p11.2) | TPR | Translocated promoter region | T-LBL, monocytosis | NA |
| Gervais et al. [90] | t(2;8)(q12;p11) | RANBP2 | RAN binding protein 2 | MDS/MPN; neutrophilia | Not tested |
| Soler et al. [91] | t(2;8)(q37;p11) | LRRFIP1 (TRIP) | LRR binding FLII interacting protein 1 | AML MRCa with eosinophilia | Not tested |
| Popovici et al. [92] | t(6;8)(q27;p11.2) | FGFR1OP | FGFR1 oncogenic partner 1 | Polycythaemia, AML, eosinophilia | Not tested |
| Belloni et al. [93] | t(7;8)(q33;p11.2) | TRIM24 | Tripartite motif containing 24 | AML, B-ALL | Not tested |
| Wasag et al. [94] | t(7;8)(q22.1;p11.2) | CUX1 | Cut like homeobox 1 | T-ALL | Sensitive to TKI258 and PKC412 |
| Guasch et al. [95]; Sohal et al. [96] | t(8;9)(p11.2;q33.2) | CNTRL (CEP110) | Centriolin | CEL, AML | Not tested |
| Sohal et al. [96] | t(8;11)(p11;p15) | NUP98 | Nucleoporin 98 | AML | Not tested |
| Sohal et al. [96] | t(8;12)(p11;q15) | CPSF6 | Cleavage and polyadenylation specific factor 6 | MPN, eosinophilia | Not tested |
| Abruzzo et al. [81] | t(8;13)(p11.2;q12.1) | ZMYM2 (ZN198) | Zinc finger MYM-type containing 2 | T-LBL, AML, eosinophilia | Ponatinib, PKC412, midostaurin |
| Walz et al. [97] | t(8;17)(p11.2;q23) | MYO18A | Myosin XVIIIA | MDS/MPN; eosinophilia, basophilia | Not tested |
| Lewis et al. [98]; Sohal et al. [96] | t(8;17)(p11.2;q25) | Not determinedb | NA | MPN, AML post mastocytosis | Not tested |
| Duckworth et al. [99] | t(8;19)(p11.2;q13.1) | ERVK-6 | Endogenous retrovirus group K member 6 | MPN with neutrophilia | Not tested |
| Montenegro-Garreaud et al. [100] | t(8;22)(p11.2;q11.2) | BCR | Breakpoint cluster region | B-ALL, monocytosis | Not tested |
| Grand et al. [101] | ins(12;8)(p11.2;p11p22) | FGFR1OP2 | FGFR1 oncogenic partner 2 | T-LBL with eosinophilia; AML | Not tested |
a AML MRC: Acute myeloid leukaemia with myelodysplasia related changes following myelodysplastic syndrome.
b Single case report in 1987; not a similar case reported up to 2017.
ALL/LBL: lymphoblastic leukaemia/lymphoma; CEL: chronic eosinophilic leukaemia; aCML: atypical chronic myeloid leukaemia.
TKI: tyrosine kinase inhibitor.
It is of interest that patients who present with acute leukaemia and receive high-dose chemotherapy may achieve complete morphological remission; however, conventional cytogenetic analysis reveals persistence of the cytogenetic abnormality, i.e. the 8p11 translocation, indicating persistent underlying chronic phase of the disease [100, 102].
Prognosis
Unlike myeloid/lymphoid neoplasms associated with PDGFRA and PDGFRB, FGFR1 rearranged neoplasms are usually not responsive to first-generation tyrosine kinase inhibitor (imatinib) therapy. Prognosis is poor and aggressive chemotherapy and haematopoietic stem cell transplantation is considered to be the best option for cure [82]. There is hope for clinical trials with third-generation tyrosine kinase inhibitors or small molecules [71, 104].
Myeloid/Lymphoid Neoplasm Associated with PCM1-JAK2 Rearrangement
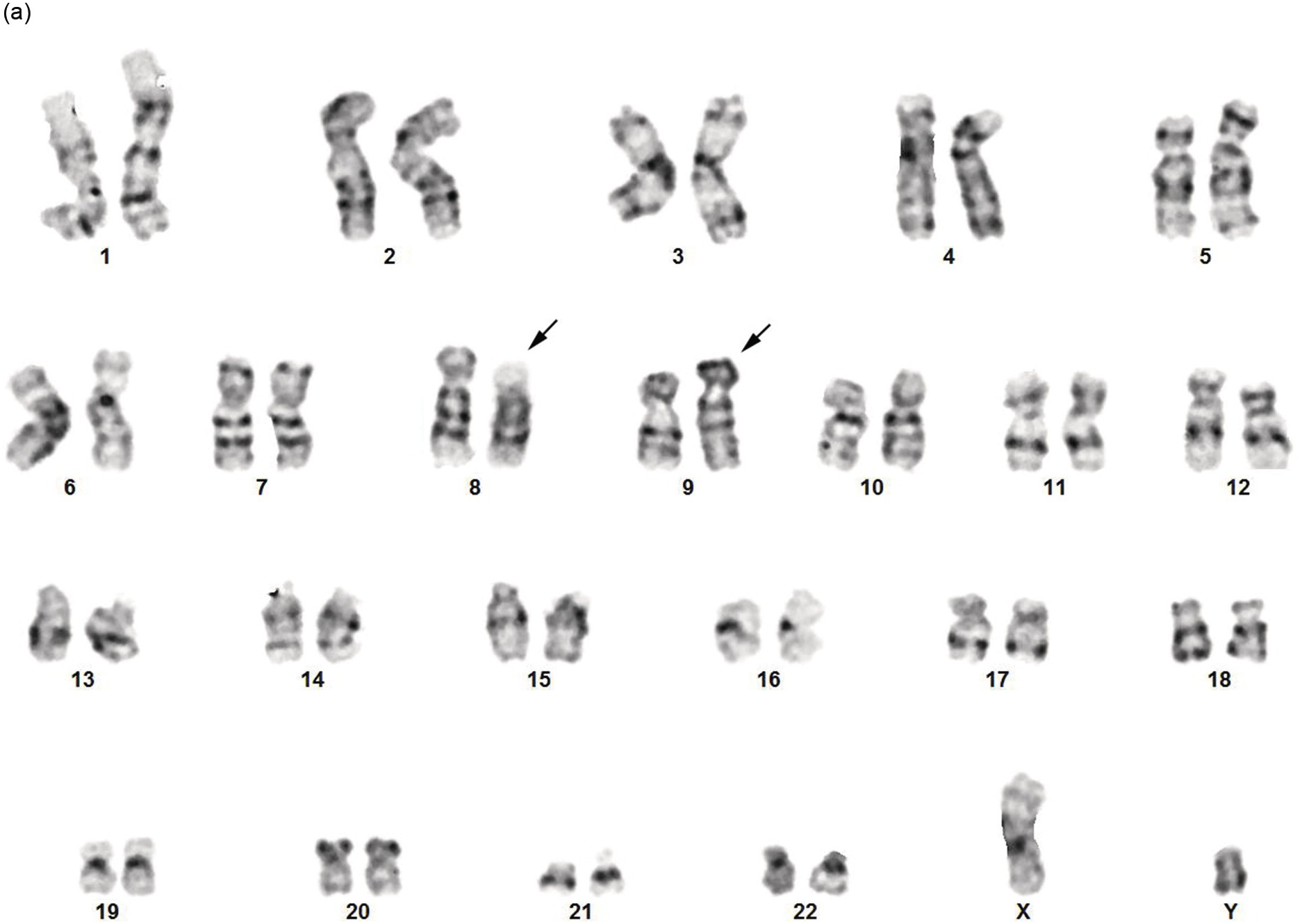
The identification of neoplasms with eosinophilia and specific tyrosine kinase rearrangements has led to increased recognition of haematolymphoid neoplasms presenting with eosinophilia but lacking PDGFRA, PDGFRB or FGFR1 rearrangements. This field will continue to evolve as new recurrent molecular genetic alterations are discovered. An example is eosinophilia associated with PCM1-JAK2 [1, 105], which has been incorporated in the group as MLNE and specific gene rearrangements as a new provisional entity in the 2016 WHO revision [1]. By conventional karyotyping, a t(8;9)(p22;p24.1) is detected, which results in a PCM1-JAK2 rearrangement.
Epidemiology
This is an extremely rare neoplasm and the geographic data is based on a summary of cases published in literature [2] that reported a median age at presentation of 47 years (range, 12 to 75 years), and a male predominance.
Clinical Features
Of the reported cases in the literature, around half were classified as an MPN and around 20% as MDS/MPN. Eosinophilia and hepatosplenomegaly are commonly associated with these cases. Basophilia can be occasionally observed. Patients may also present with AML, B-ALL, T-ALL/LBL or peripheral T-cell lymphoma.
Morphology
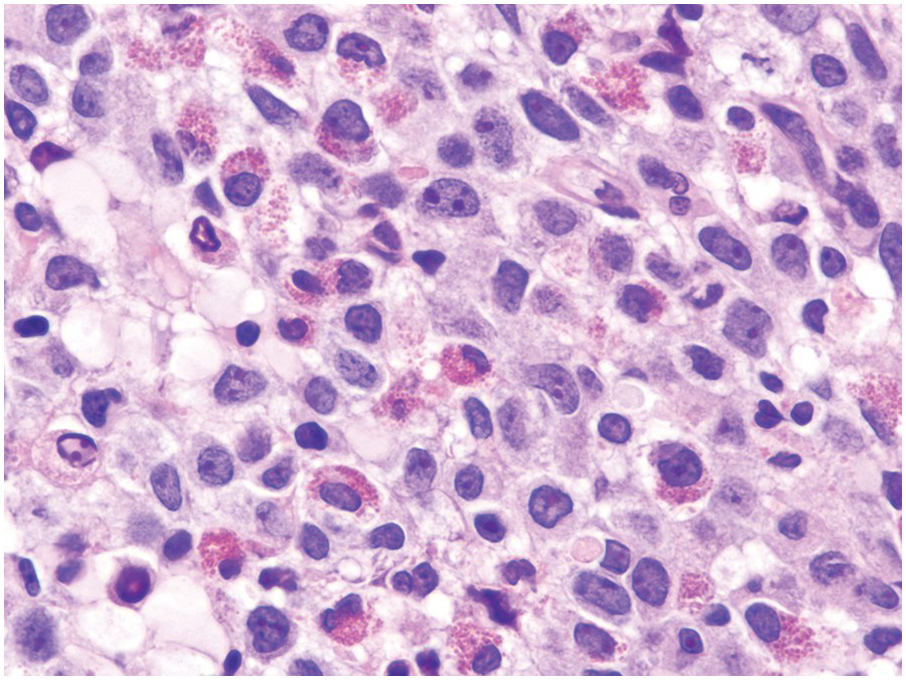
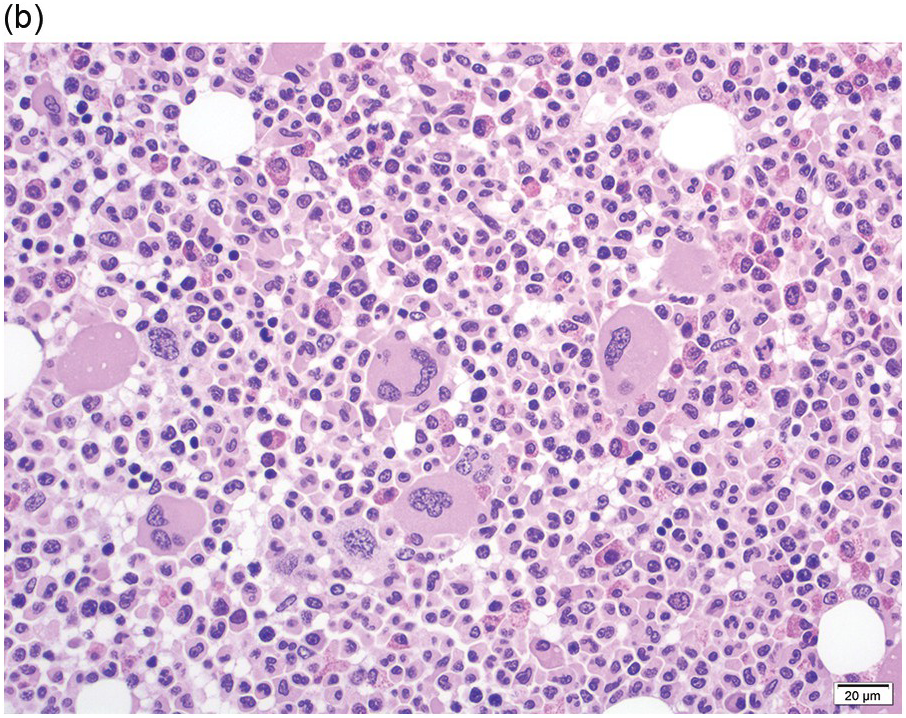
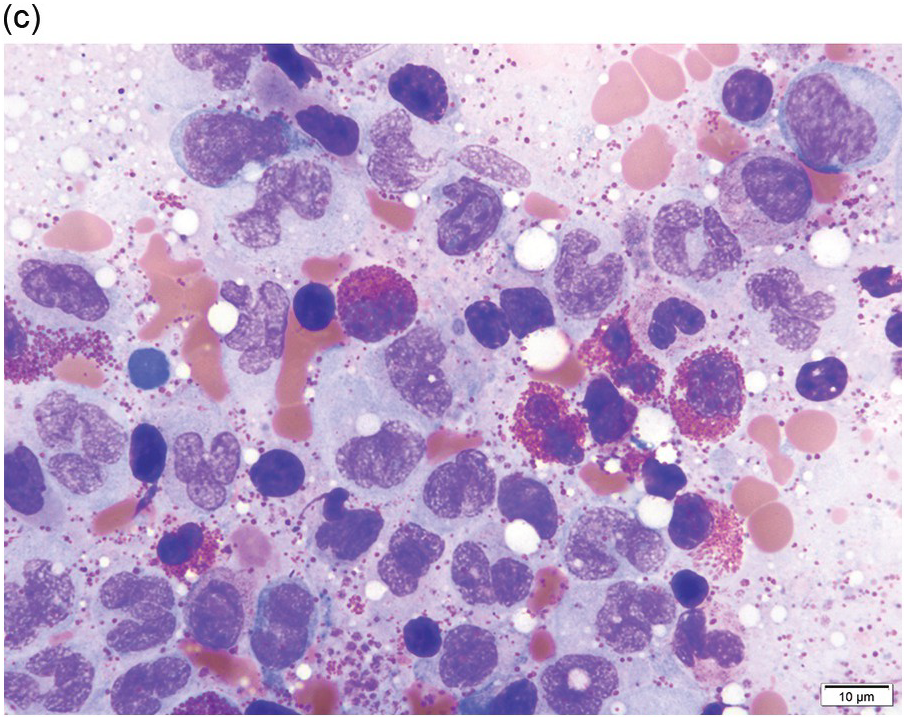
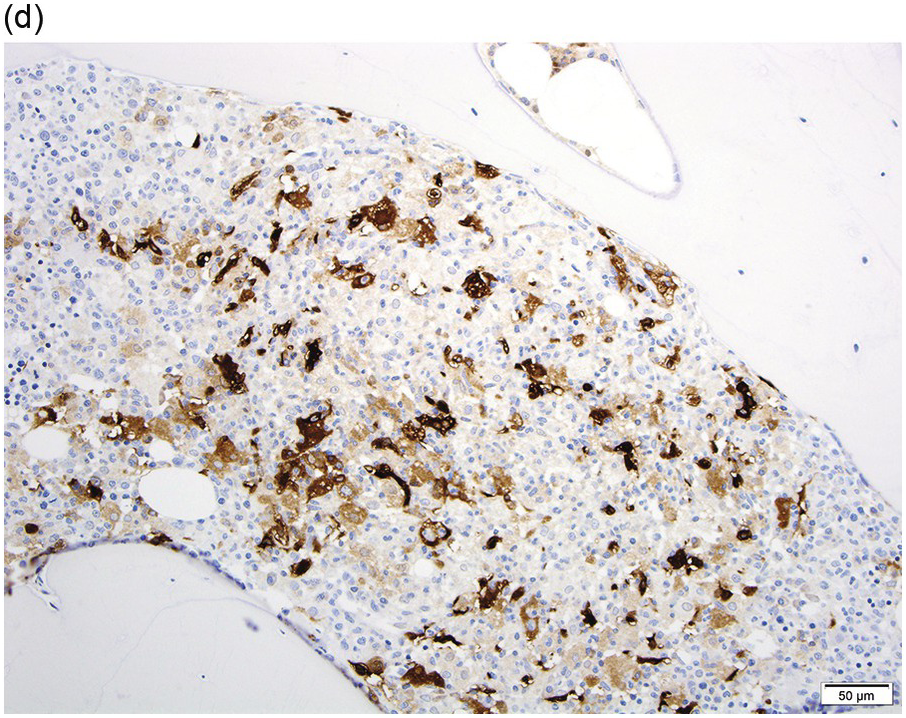
Prior to the recognition of these neoplasms as a provisional entity [1], the cases with a chronic presentation were diagnosed as CEL, NOS, primary myelofibrosis (PMF), atypical CML, MPN-unclassifiable or MDS/MPN-unclassifiable, indicating the morphologic resemblance to these neoplasms. However, some unique morphological features have been described. The BM biopsy (BMB) (Figure 13.8) shows hypercellularity with increased eosinophils, increased immature erythroid precursors (pronormoblasts) in aggregates or sheets, and myelofibrosis. Erythroid hyperplasia and prominent dyserythropoiesis may be present. Lymphoid aggregates may also be present. Some cases show dysgranulopoiesis. Monocytosis is uncommon. Cases presenting as AML with PCM1-JAK2, were formerly classified as AML without maturation, AML with maturation, erythroleukaemia or AML progression from an underlying (chronic) MPN or MDS/MPN.
(a) Bone marrow biopsy shows hypercellularity.
(b) Higher magnification of the BMB shows a large cluster of erythroid precursors in a background of neutrophils and eosinophils.
(c) High magnification of the immature cell cluster demonstrates that most cells are erythroid precursors.
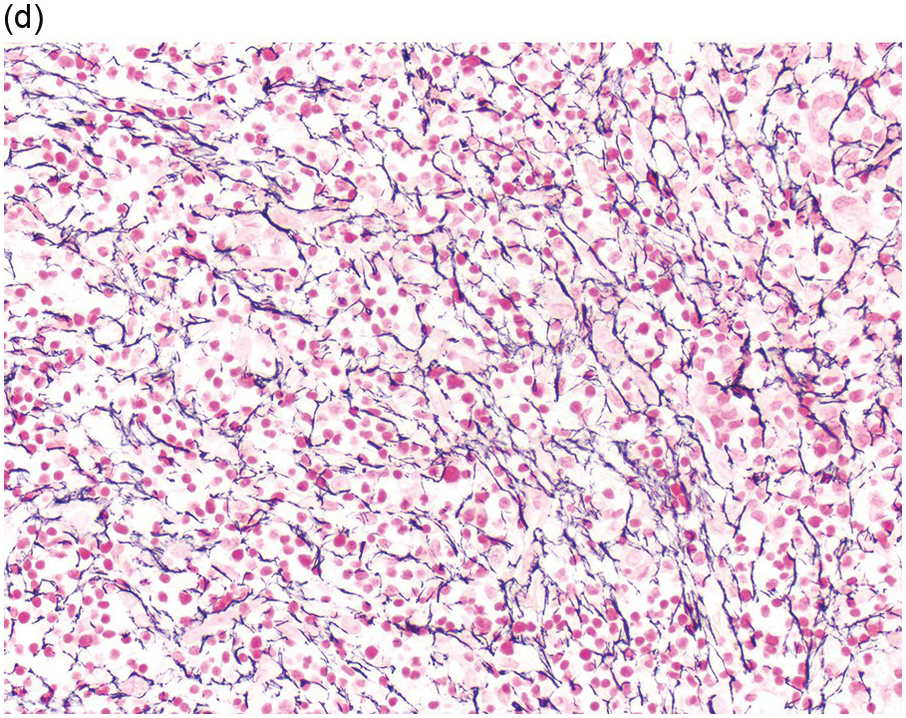
(d) Reticulin stain highlights increased reticulin fibrosis. These are characteristic features of PCM1-JAK2.
Immunophenotype
If blasts are increased, a comprehensive immunophenotyping study should be performed to characterize the blasts. Immature erythroid precursors can be confirmed by CD71 and E-cadherin immunohistochemistry studies.
Cytogenetics and Molecular Genetics
The t(8;9) (p22;p24.1) with PCM1-JAK2 rearrangement should be distinguished from t(8;9)(p11;q33)/CEP110-FGFR1. FISH for JAK2 and FGFR1 molecular abnormalities should be performed to confirm the rearrangement suggested by the karyotyping study (Figure 13.9). For cases presenting with B-ALL without an associated underlying MPN, it is not currently clear whether these cases should be more appropriately classified as BCR-ABL1–like B-ALL.