Abstract
Mastocytosis is a diverse group of rare diseases due to a clonal proliferation of neoplastic mast cells that can involve a wide variety of organ systems. The two main categories of mastocytosis are cutaneous mastocytosis (CM) showing only skin involvement, and systemic mastocytosis (SM) with at least one extracutaneous organ involved. In many cases of SM, the bone marrow (BM) shows varying degrees of infiltration. Most cases of CM develop during childhood, while adult patients in their fifth and sixth decades tend to present with SM [1]. The clinical course can vary from spontaneous regression in young children with CM to a highly aggressive course primarily seen in adult patients. Even within the category of SM, the presentation can range from indolent to aggressive, and it is thus divided into five subcategories as outlined in the most recent edition of the 2016 World Health Organization (WHO) classification of mastocytosis (Table 12.1) [2]. Of note, mastocytosis is now considered a distinct clinicopathologic entity that is separate from other myeloproliferative neoplasms.
Introduction
Mastocytosis is a diverse group of rare diseases due to a clonal proliferation of neoplastic mast cells that can involve a wide variety of organ systems. The two main categories of mastocytosis are cutaneous mastocytosis (CM) showing only skin involvement, and systemic mastocytosis (SM) with at least one extracutaneous organ involved. In many cases of SM, the bone marrow (BM) shows varying degrees of infiltration. Most cases of CM develop during childhood, while adult patients in their fifth and sixth decades tend to present with SM [1]. The clinical course can vary from spontaneous regression in young children with CM to a highly aggressive course primarily seen in adult patients. Even within the category of SM, the presentation can range from indolent to aggressive, and it is thus divided into five subcategories as outlined in the most recent edition of the 2016 World Health Organization (WHO) classification of mastocytosis (Table 12.1) [2]. Of note, mastocytosis is now considered a distinct clinicopathologic entity that is separate from other myeloproliferative neoplasms.
| Indolent systemic mastocytosis |
| Smouldering systemic mastocytosis |
| Systemic mastocytosis with an associated haematologic neoplasma |
| Aggressive systemic mastocytosis |
| Mast cell leukaemia |
a Previously termed systemic mastocytosis with an associated clonal haematological non-mast cell lineage disease (SM-AHNMD).
SM is defined as involvement of at least one extracutaneous organ, with or without cutaneous involvement [2]. A diagnosis of SM must fulfil the major criterion in addition to one minor, or three minor criteria by themselves (Table 12.2). The major criterion may not always be present; therefore, additional ancillary testing, such as flow cytometric analysis, immunohistochemistry and molecular studies, is typically performed.
| Major criteria: |
| Multifocal dense infiltrates of mast cells (>15 mast cells in aggregate)a |
| Minor criteria: |
| 1. More than 25% of the mast cells are spindle-shaped or have atypical morphologyb |
| 2. Expression of CD25 with or without CD2c |
| 3. Detection of activating point mutation at codon 816 of KITd |
| 4. Serum tryptase level persistently elevated >20 ng/mLe |
a Aggregates of mast cells can either be detected in the bone marrow or tissue sections of extracutaneous organs.
b While the majority of cases of systemic mastocytosis show atypical or spindle-shaped mast cells, some cases can show more mature well-differentiated morphology.
c CD25 expression is a more sensitive marker by both flow cytometry and immunohistochemistry.
d Either detected from a bone marrow or peripheral blood sample or tissue from an extracutaneous organ.
e Unable to use as a minor criterion if the patient has an associated myeloid neoplasm, which can also cause elevated tryptase levels.
Postulated Cell of Origin
Mast cells were discovered over 100 years ago by Paul Ehrlich who applied dyes to demonstrate metachromasia in the cytoplasmic granules. This observation earned Ehrlich the Noble Prize in Medicine in 1908 [3]. The cytoplasmic granules aid in the initial identification of mast cells in tissues, although in advanced forms of SM morphologic identification of mast cells can be difficult.
Mast cells are derived from haematopoietic stem cells that differentiate and mature via different cytokines and other growth factors. Mast cell derived progenitor cells express the KIT receptor that is stimulated by a cytokine called stem cell factor (SCF), also known as KIT-ligand or mast cell growth factor. Stem cell factor is the major growth factor responsible for the differentiation of mast cells [4, 5]. Mast cells continue their maturation process and serve multiple purposes within various tissue types. Normal proportions of mast cells vary greatly depending on the tissue of interest [6].
Mast cells are effector cells that are involved in many different functions including regulation of inflammation and tissue remodeling, collagen synthesis, immune reactions and angiogenesis, among others [6, 7]. The most recognized activation pathway of mast cells occurs via binding of the FcɛRI receptor on the cell surface to an IgE immunoglobulin [8]. Activation can also occur via other receptor mediated or non-receptor mediated pathways. Once activated, mast cells release their granule contents; this can occur during inflammation, tumour growth, immune-mediated reactions, and exposures to certain drugs and/or toxins [6]. The contents within mast cell granules include heparin, histamine, proteases, tryptases, chymases and other biologically active substances [4]. The release of these factors leads to the various clinical signs and symptoms ranging from mild to life-threatening anaphylaxis. Interestingly, mast cells are considered long-lived cells that are capable of a regranulation process [4].
Systemic Mastocytosis
Morphologic review of the BM is an important diagnostic step in the evaluation of SM. BM aspirates typically contain only rare mast cells due to accompanying fibrosis, variability in the number of mast cells present and the location of the mast cell aggregates. Mast cells tend to localize in paratrabecular, perivascular and perisinusoidal areas. Paratrabecular infiltrates often cause osteosclerosis of the bony trabeculae [1]. Determination of the degree of BM involvement by SM should be based on a tryptase-stained BM biopsy (BMB) section [9].
There are many well-characterized morphologic subtypes of mast cells that may be seen in SM (Figure 12.1). The mature-appearing mast cell has well-granulated cytoplasm and a centrally placed nucleus; well-differentiated mast cells show similar features, although they tend to be larger in size. The atypical mast cell type 1 is spindle-shaped. The atypical mast cell type 2 is also referred to as a promastocyte with features including bi- or multinucleation. Finally, the metachromatic blast is immature with fine chromatin detail and often contains hypogranulated cytoplasm [1].
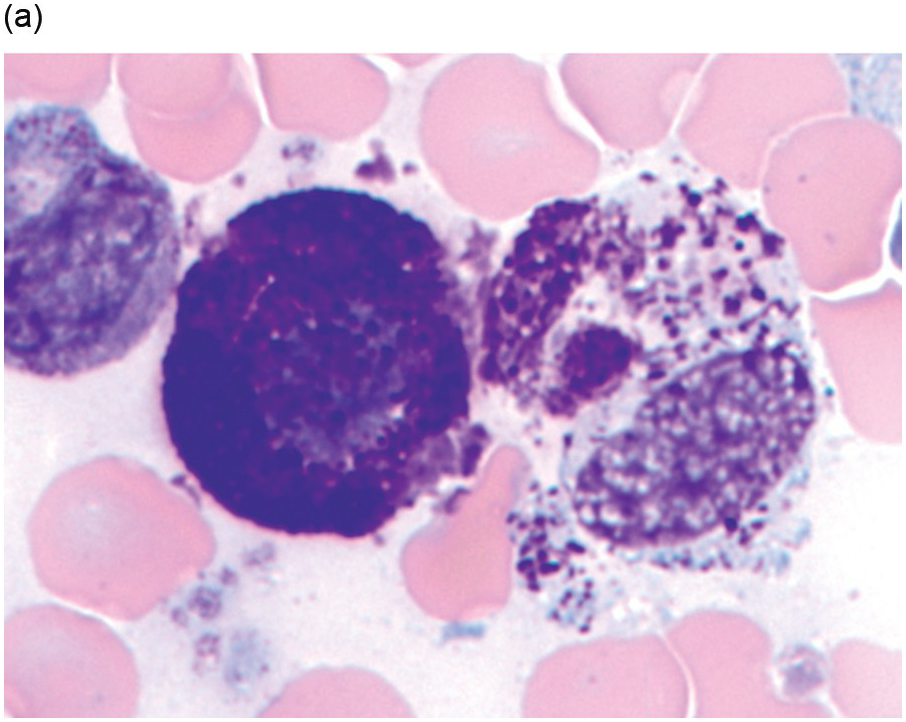
(a) In this patient with indolent systemic mastocytosis, a normal round mast cell is present with cytoplasm containing numerous metachromatic granules, while on the right, a spindle-shaped mast cell is present. The neoplastic mast cell is hypogranular when compared to the normal mast cell and it contains elongated cytoplasmic projections with an oval-shaped nucleus.
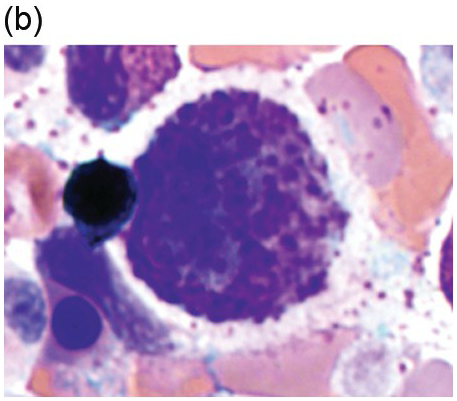
(b) A promastocyte is present from this patient with acute mast cell leukaemia. The immature mast cell is round with a nucleus containing irregular nuclear contours. The cytoplasm is hypogranular with scattered metachromatic granules and the chromatin is partially condensed. A background eosinophilia is present.
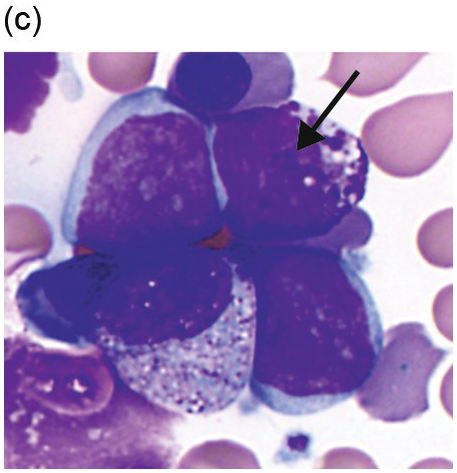
(c) An immature mast cell or metachromatic blast is present (arrow) with a blastic nucleus and several large metachromatic granules. Two adjacent myeloblasts are also present in this patient with myelomastocytic leukaemia. (Bone marrow aspirate smears, Wright–Giemsa stain.)
Figure 12.1 Cytomorphology of mast cells.
Advanced mastocytosis includes the following categories: aggressive systemic mastocytosis (ASM), systemic mastocytosis with an associated haematologic neoplasm (SM-AHN) and mast cell leukaemia (MCL). More indolent subtypes include indolent systemic mastocytosis (ISM) and smouldering systemic mastocytosis (SSM). Each of the following subtypes of SM will be discussed separately below as the clinical, pathologic findings and prognostic differences vary greatly.
Indolent Systemic Mastocytosis
Indolent systemic mastocytosis (ISM), as the name suggests, is an indolent subtype of SM that almost invariably shows cutaneous involvement. The diagnosis of ISM is one in which the criteria are met for SM as discussed in Table 12.2. A patient may display up to one B finding, but if two or more B findings are present then the patient would be in the now separate category of SSM (Table 12.3). In addition, patients must not display any C findings or have involvement by another haematologic neoplasm (Table 12.4).
Table 12.3 Burden of disease findings.
| ‘B’ findings: |
| 1. High mast cell burden with >30% bone marrow infiltration and serum tryptase >20 ng/mL |
| 2. Presence of myelodysplasia or myeloproliferation in non-mast cell lineages, but not meeting the criteria for an associated haematologic neoplasm |
| 3. Organomegaly: hepatomegaly, lymphadenopathy and/or splenomegaly; without impaired function |
Table 12.4 Cytoreductive findings.
| ‘C’ findings: |
| 1. Bone marrow dysfunction due to neoplastic mast cell infiltration, defined as ≥1 cytopaenia: |
| a. Absolute neutropaenia <1 × 109/L |
| b. Haemoglobin <10 g/dL |
| c. Platelet count <100 × 109/L |
| 2. Hepatomegaly with impaired liver function, ascites and/or portal hypertension |
| 3. Skeletal involvement with large osteolytic lesions |
| 4. Splenomegaly with hypersplenism |
| 5. Malabsorption with weight loss due to gastrointestinal mast cell infiltrates |
Synonyms
None.
Epidemiology
Of all the subtypes of SM, ISM is the most commonly diagnosed entity (approximately 50% of cases) and usually presents in adulthood [5, 10]. Increased detection and awareness of mastocytosis in general are likely to uncover even more patients with ISM.
Clinical and Laboratory Features
Signs and symptoms of ISM are often related to mast cell degranulation and their associated mediator effects produced by the various substances within the granules. Most patients show skin manifestations typical of urticaria pigmentosa (UP), which is now known as maculopapular cutaneous mastocytosis (MPCM); this is present in more than 90% of cases [10]. However, other less common cutaneous lesions may also be seen. Organomegaly or lymphadenopathy are much less common in ISM compared to other subtypes of SM [11]. The serum tryptase level is often only mildly elevated over 20 ng/mL. The KIT D816V mutation is found in >90% of cases as well [2].
Morphology
The BM shows dense, multifocal aggregates of mast cells that are sharply demarcated from residual haematopoiesis. The overall mast cell burden is low with paratrabecular and perisinusoidal mast cell aggregates, often admixed or adjacent to benign lymphoid aggregates, polyclonal plasma cells and eosinophils (Figure 12.2). Mast cells morphologically may appear mature and round or may be more atypical with either spindle-shaped, hypogranulated cytoplasm or eccentric nuclei. Occasionally, mast cell infiltrates may be present within other organs such as the gastrointestinal system, liver, etc. Areas of osteosclerosis are typically focal and adjacent to the mast cell aggregates.
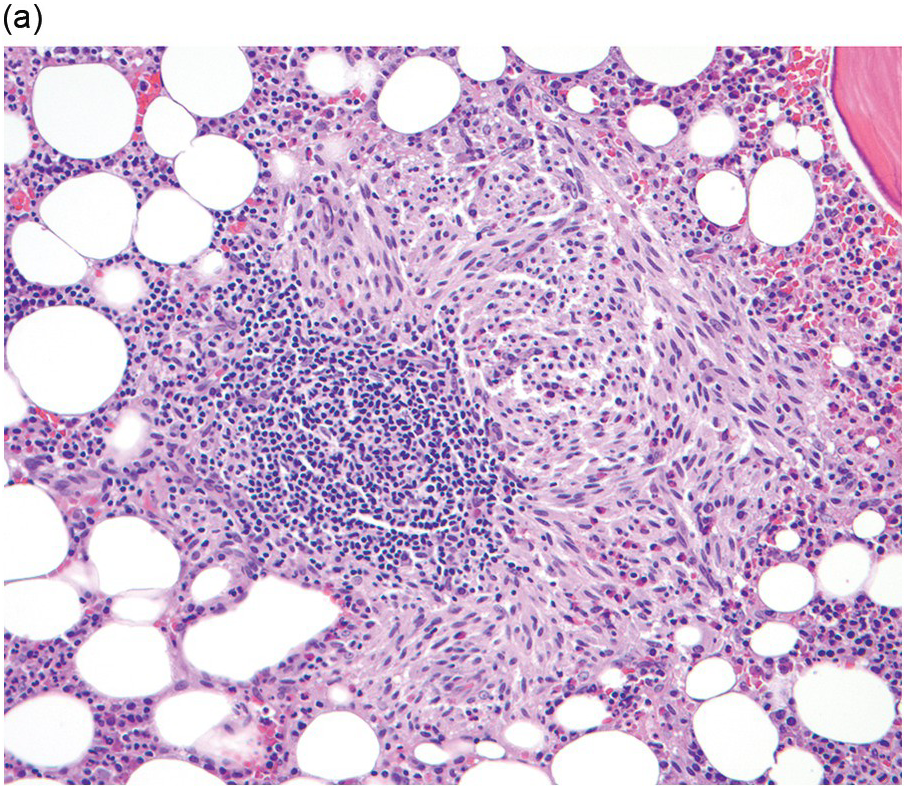
(a) A dense aggregate of spindle-shaped mast cells is present surrounding a reactive lymphoid aggregate. The mast cells are admixed with scattered eosinophils, which are prominent at the edge of the mast cell aggregate.
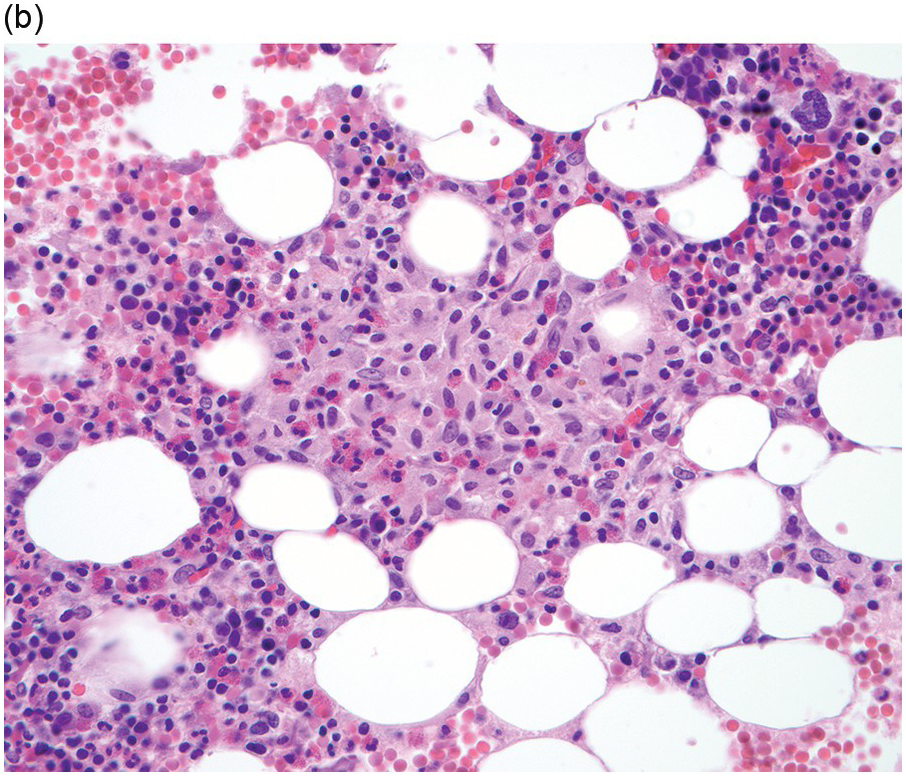
(b) A closer examination of this mast cell nodule shows the admixture of spindle-shaped and round mast cells with abundant lightly eosinophilic cytoplasm and oval nuclei lacking prominent nucleoli. Scattered mature eosinophils are present. (Bone marrow trephines, H&E.)
Figure 12.2 Indolent systemic mastocytosis.
Subtype
Isolated bone marrow mastocytosis (BMM) represents a rare subtype of ISM. These patients present with only BM mast cell involvement and show no cutaneous lesions or extramedullary organ involvement. The mast cell burden is typically low and serum tryptase levels are also near the normal range [2]. Of note, more aggressive subtypes of SM may also lack skin lesions, but tend to show signs and symptoms of SM correlating with a higher mast cell burden and significantly elevated serum tryptase levels.
Prognosis and Treatment
Patients with ISM tend to have an overall excellent prognosis compared to other subtypes of SM. Transformation to more aggressive mastocytosis or leukaemia can rarely occur. Treatment therefore is aimed at symptomatic relief from mediator related symptoms and avoidance of triggers, including the use of histamine receptor antagonists [11]. Cytoreductive therapy is typically avoided unless the patient is refractory to treatment [11].
Smouldering Systemic Mastocytosis
Smouldering systemic mastocytosis (SSM) was once considered a subtype of ISM, but it is now a separate variant of SM according to the WHO 2016 classification [2]. SSM fulfils the diagnostic criteria for SM with the addition of two or more B findings. Patients may not have any C findings or other associated haematologic neoplasm to be in this category.
Synonyms
None.
Epidemiology
Exact numbers are unknown at this time as SSM is now considered a new entity that was previously subclassified under ISM.
Clinical and Laboratory Features
Serum tryptase levels are usually high with values >200 ng/mL [5]. In most cases, the KIT D816V mutation is present not only in mast cells, but can also be identified in other myeloid cells [2].
Morphology
The mast cell burden is typically high in the BM with not only dense multifocal infiltrates, but areas of diffuse infiltrates of mast cells are also present [12]. Aspirate smears, however, show less than 20% mast cells by differential count. In addition to the high mast cell burden, the marrow is often hypercellular with loss of fat cells. There may be signs of accompanying dysplasia or myeloproliferation as well, but overall criteria are not met for a myelodysplastic or myeloproliferative neoplasm. Mast cell morphology may vary from slightly atypical to highly atypical; however, cells that are highly atypical tend to be in the minority [9].
Prognosis and Treatment
The prognosis of patients with SSM is less favourable compared to ISM, but more favourable than advanced forms of SM [13]. These patients should be more closely monitored as progression to ASM or MCL is more likely to occur compared to ISM [2]. Treatment may involve just a ‘watch and wait’ approach while monitoring tryptase levels and clinical follow-up [11]. Therefore, treatment is quite similar to ISM with avoidance of triggers and reduction in mediator related symptoms. In patients that show progression in B findings a cytoreductive agent may be added to the therapy regimen [11].
Systemic Mastocytosis with an Associated Haematologic Neoplasm
Systemic mastocytosis with an associated haematologic neoplasm (SM-AHN) is a category in which the criteria for SM are met along with a recognized haematologic neoplasm according to the WHO classification. These patients must be categorized not only for the type of haematologic neoplasm present, but also the type of SM identified for therapeutic and prognostic implications, if possible.
The type of associated haematologic neoplasm in most cases is of myeloid lineage such as a myeloproliferative neoplasm (MPN), myelodysplastic syndrome (MDS), MPN/MDS such as chronic myelomonocytic leukaemia, or acute myeloid leukaemia (AML) (Figure 12.3). Rarely, non-Hodgkin lymphoma and plasma cell myeloma may be present, but these are not typically clonally related to the SM present [1, 5]. Of note, the most commonly detected non-mast cell neoplasm is an MDS/MPN [2].
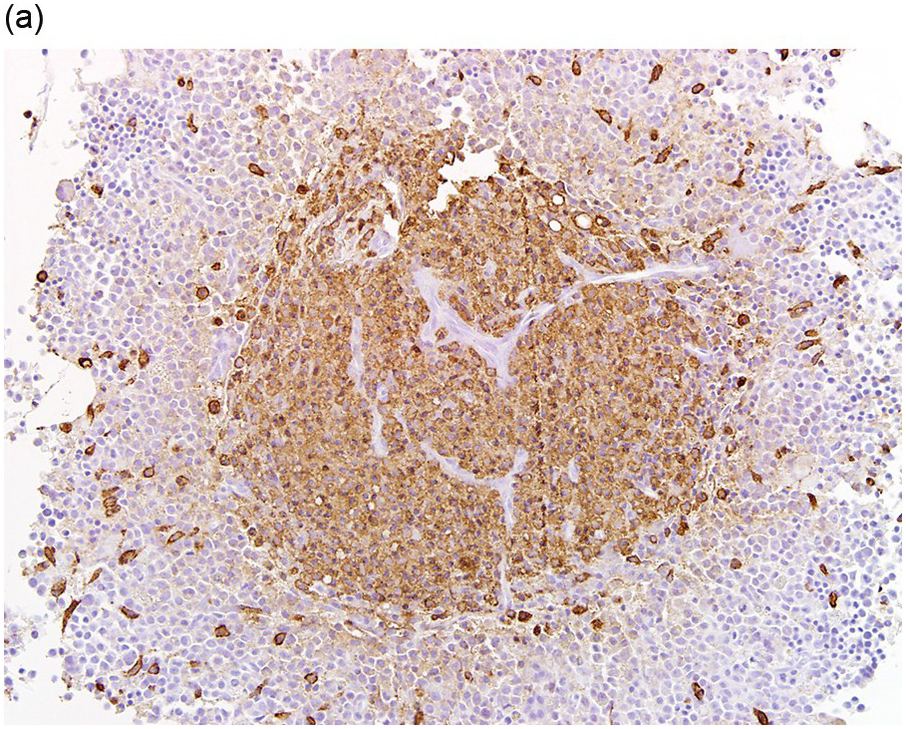
(a) Immunohistochemical stain for tryptase highlights this dense mast cell aggregate.
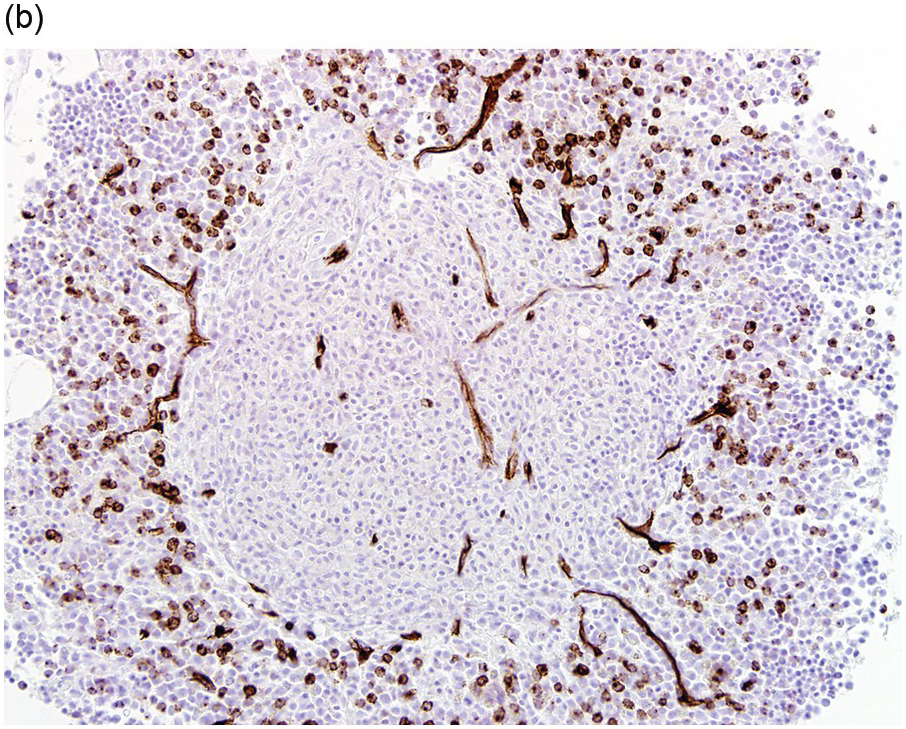
(b) The mast cells lack expression for CD34, but increased numbers of CD34-positive myeloblasts are present surrounding the nodule of mast cells.
Figure 12.3 Systemic mastocytosis with an associated haematological neoplasm. This 77-year-old male was noted to have systemic mastocytosis and myelodysplastic syndrome with excess blasts.
The SM component can vary from ISM to MCL; however, not uncommonly, the diagnosis of an underlying SM can be difficult to appreciate initially if the associated haematologic neoplasm is predominant. In other instances, the opposite may be true with an obscured non-mast cell haematologic neoplasm by advanced mastocytosis. In the latter situation, often the SM is revealed after therapy for the non-mast cell haematologic malignancy [14].
Synonyms
Previously referred to systemic mastocytosis with an associated clonal haematologic non-mast cell lineage disease SM-AHNMD.
Epidemiology
Systemic mastocytosis with an associated haematologic neoplasm is the second most common type of SM identified after ISM [1]. The true incidence in the literature varies from less than 5% up to 40% of patients with SM [10, 15]. The degree of discrepancy can be explained by the difficulty in detecting a concurrent SM component in the overwhelming presence of another haematologic neoplasm and rarely vice versa. Patients tend to be older compared to those without a concurrent haematologic neoplasm [15].
Clinical and Laboratory Features
Abnormalities such as anaemia, thrombocytopaenia and/or leukocytosis are often present. The serum tryptase level is usually >20 ng/mL, but cannot be evaluated as a minor criterion if there is another myeloid neoplasm; this is because other myeloid neoplasms can express tryptase. Constitutional symptoms, hepatosplenomegaly and lymphadenopathy are much more frequently detected than in patients with ISM [15].
Not only is the KIT D816V mutation identified in the vast majority of cases, but additional clonal abnormalities are often detected by conventional cytogenetics, FISH and/or molecular genetic analysis. The most frequently detected abnormalities identified by next-generation sequencing include mutations in TET2, SRSF2, ASXL1, CBL and EZH2. These mutations are not specific to mastocytosis, though, and can be seen in other myeloid neoplasms. The sequence of molecular events shows that in cases of advanced SM the D816V KIT mutation is a late acquired event [16].
Morphology
Typically, the BM is hypercellular with multifocal compact aggregates of mast cells. However, the degree to which mast cells are identified largely depends on the type of non-mast cell neoplasm present. Care should be taken when evaluating cases that normally may show increased numbers of mast cells, such as in lymphoplasmacytic lymphoma, MPN with eosinophilia exhibiting PDGFRA, PDGFRB or FGFR1 mutations, hypereosinophilic syndrome (HES), chronic eosinophilic leukaemia (CEL), etc. (Figure 12.4). These cases must show multifocal compact aggregates of mast cells fulfilling the major criteria plus one minor criterion or three minor criteria to allow a diagnosis of SM-AHN [1].
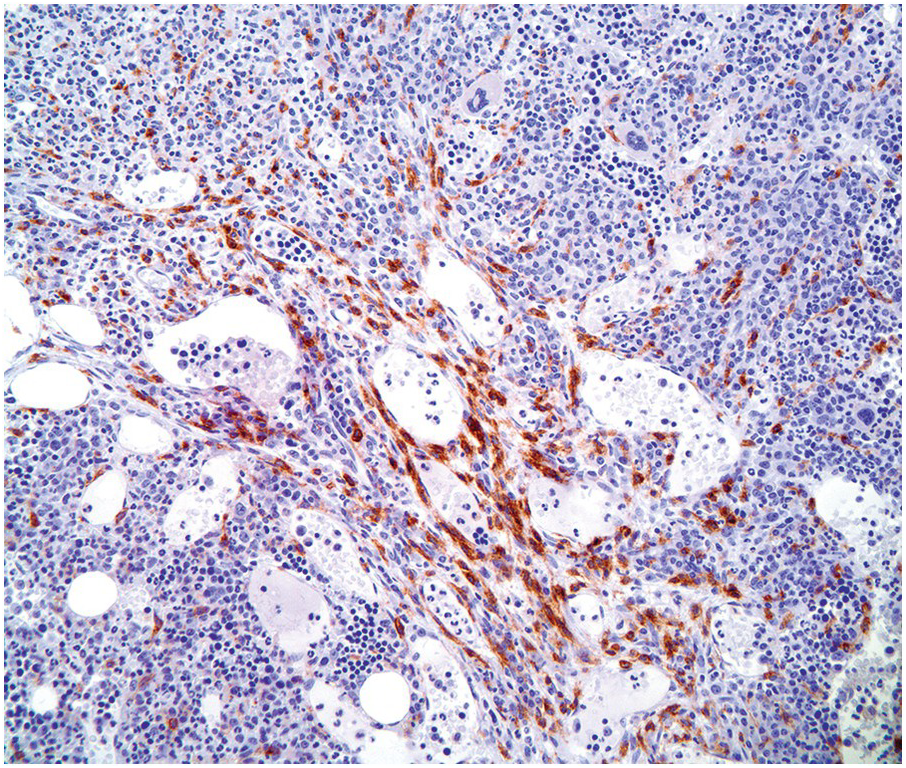
Figure 12.4 Chronic eosinophilic leukaemia with FIP1L1-PDGFRA. Increased spindle-shaped mast cells are present in the bone marrow biopsy of this patient with PDGFRA-mutated chronic eosinophilic leukaemia. The CD25-positive mast cells are concentrated around marrow sinuses but the dense compact aggregates of systemic mastocytosis are not seen. (Bone marrow biopsy, CD25 immunohistochemical stain.)
Prognosis and Treatment
The overall median survival for patients with SM-AHN is two years, which is significantly less than ISM with more than 15 years expected survival [10]. The prognosis is largely based on the type of associated haematologic neoplasm present. Treatment plans are typically separate with regimens targeted at both the SM and the associated haematologic neoplasm components [11].
Aggressive Systemic Mastocytosis
Aggressive systemic mastocytosis (ASM) is a rare subtype of SM that fulfils the diagnosis of SM with one or more C findings. In contrast to MCL, the mast cells account for less than 20% of all nucleated BM cells [2].
Synonyms
None.
Epidemiology
The exact incidence is unknown but is thought to represent less than 5% of all cases of SM [1].
Clinical and Laboratory Features
The mast cell infiltrates in ASM cause severe organ impairment within the liver, spleen, BM and skeletal systems [5]. Patients may present with pathologic bone fractures. Skin findings are typically absent. Laboratory evaluation often shows a markedly elevated serum tryptase level of >20 ng/mL [10]. Additional clonal abnormalities may be detected, but at a lower frequency compared to more indolent subtypes and SM-AHN.
Morphology
The BM is hypercellular and often shows a mixture of multifocal compact aggregates of mast cells and areas of diffuse mast cell infiltrates [12] (Figure 12.5). Mast cells are often atypical in appearance with hypogranular forms and spindle-shaped cells [1].
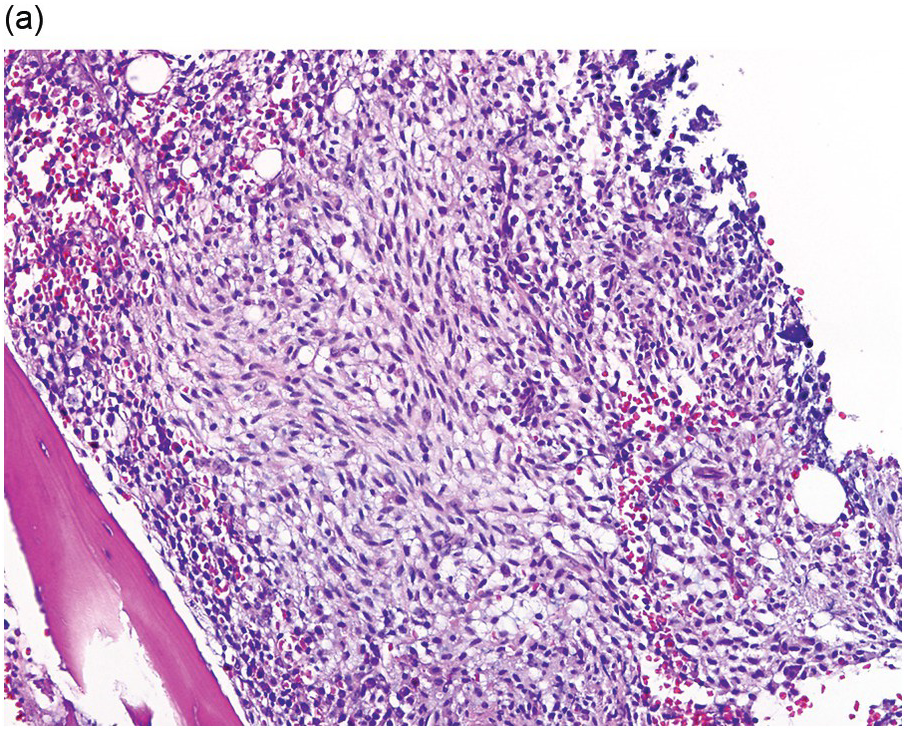
(a) The bone marrow was almost entirely replaced by aggregates and sheets of mast cells as shown in this image with predominantly spindle-shaped mast cells. (Bone marrow trephine, H&E.)
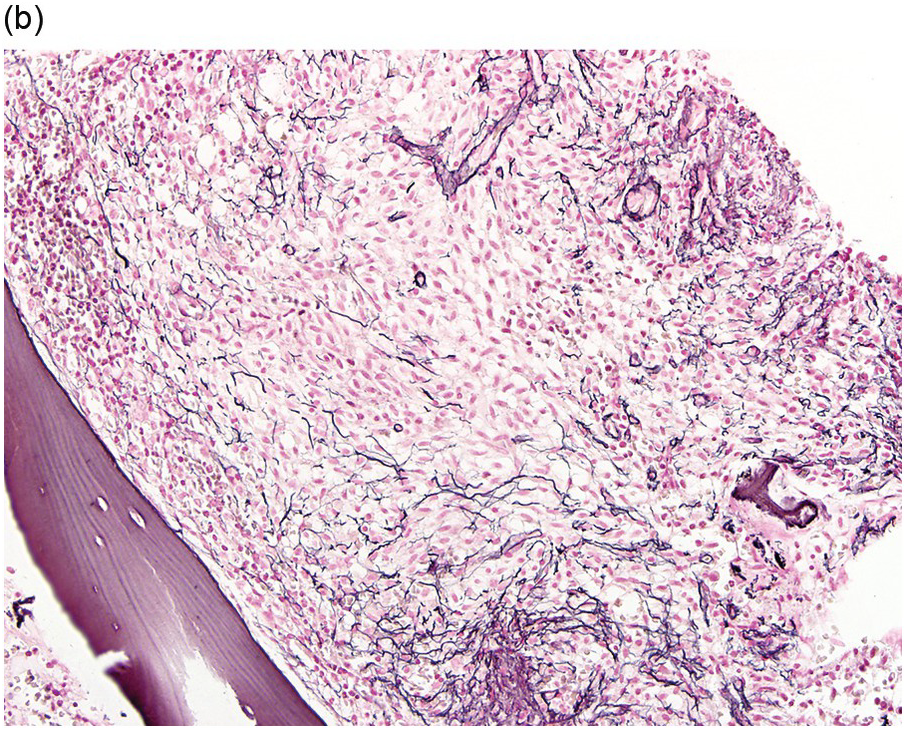
(b) A reticulin stain highlights the increased fibrosis surrounding the mast cell aggregate. (Bone marrow trephine, reticulin stain.)
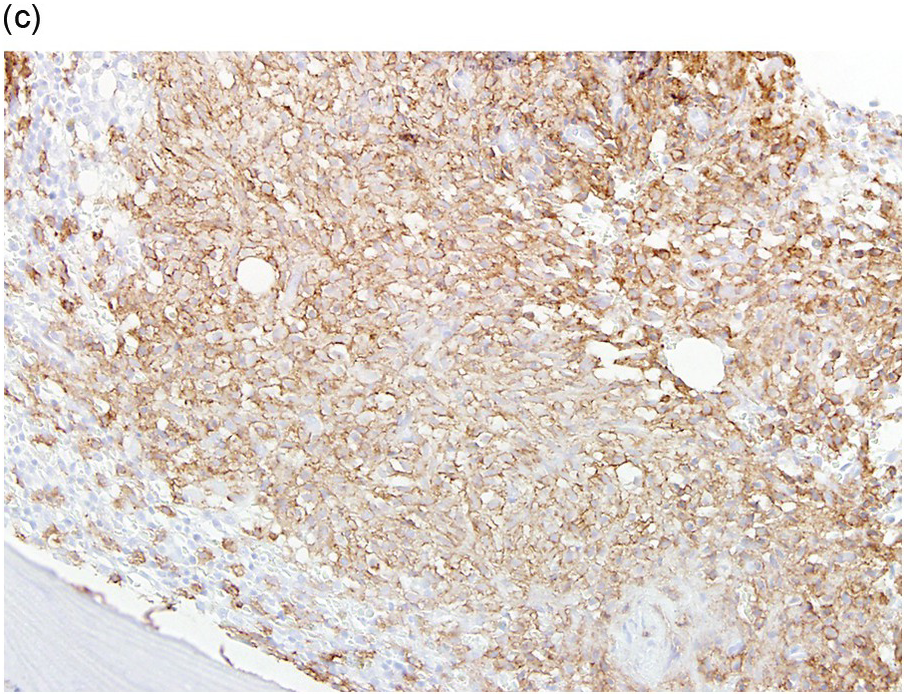
(c) Immunohistochemical stain for CD117 (KIT) uniformly highlights the mast cells with membranous staining. (Bone marrow trephine, CD117.)
Figure 12.5 Aggressive systemic mastocytosis.
Subtypes
According to the WHO 2016 classification, ASM is now divided into untransformed and transformed variants, also known as ‘ASM in transformation to MCL’. The distinction relies upon the percentage of mast cells in BM aspirate smears [13]. Patients with ASM in transformation (ASM-t) show greater than 5%, but fewer than 20% mast cells in BM aspirate smears [2]. This distinction is important as patients identified as ASM-t are likely to evolve into MCL in a relatively short timeframe of only a few months (Figure 12.6) [13].

Figure 12.6 Aggressive systemic mastocytosis transformed to mast cell leukaemia. Sheets of well granulated round mast cells replace the bone marrow on this Wright–Giemsa-stained aspirate smear.
Prognosis and Treatment
The prognosis for patients with ASM is poor with an average median survival of three years [10]. Patients with ASM-t have been identified to show an even worse prognosis, as mentioned above. Responses to various cytoreductive treatments are suboptimal in most cases. For patients that can tolerate it and have a suitable donor, stem cell transplantation is a viable option to provide a chance at remission [17].
Mast Cell Leukaemia
Mast cell leukaemia (MCL) is also another rare subtype of SM that is a highly malignant neoplasm. By definition, more than 20% mast cells are present on aspirate smears. In most cases, criteria for SM are met. The majority of MCL arise de novo and are termed primary MCL with no known preexisting mast cell neoplasm; however, cases of secondary MCL can also occur [18].
Synonyms
The aleukaemic variant of MCL was previously termed malignant mastocytosis with circulating mast cells.
Epidemiology
The diagnosis of MCL is extremely uncommon, comprising <1% of cases of SM. Interestingly, most patients are female, compared to other subtypes of SM with a slight male predominance. Most patients are in their fifth decade of life, while those with secondary MCL present earlier in their thirties [18]. Of the few cases reported in the literature, no familial cases have been reported.
Clinical and Laboratory Features
Patients with MCL usually show a rapid and progressive clinical course. C findings are often present and cause significant morbidity and mortality, in particular prominent cytopaenias (Table 12.4). Clinical symptoms related to mast cell mediator effects are also present, but skin findings are characteristically absent [10]. Tryptase levels are often exceedingly elevated, which is indicative of the high mast cell burden. The KIT D816V mutation is detected in a portion of patients, while in other patients a non-D816V mutation within exon 17 or other mutations within the KIT gene may be present [18]. Importantly, juxtamembrane and other mutations of KIT may be sensitive to imatinib. Therefore gene sequencing of the KIT gene may be considered in patients with MCL for more definitive evaluation.
Morphology
All subtypes of MCL show greater than 20% mast cells in the BM aspirate smears. Enumeration of mast cells on BM aspirate smears should be counted away from marrow spicules [19]. The BM is hypercellular with loss of normal haematopoiesis due to the diffuse and compact pattern of infiltration of mast cells [19]. Mast cells often show atypical morphology including metachromatic blasts with immature blast-like morphology and hypogranulated cytoplasm [19]. In addition, some immature mast cells may have irregular nuclear contours, or even bi- or multilobed nuclei, referred to as promastocytes (Figure 12.7) [5, 19].
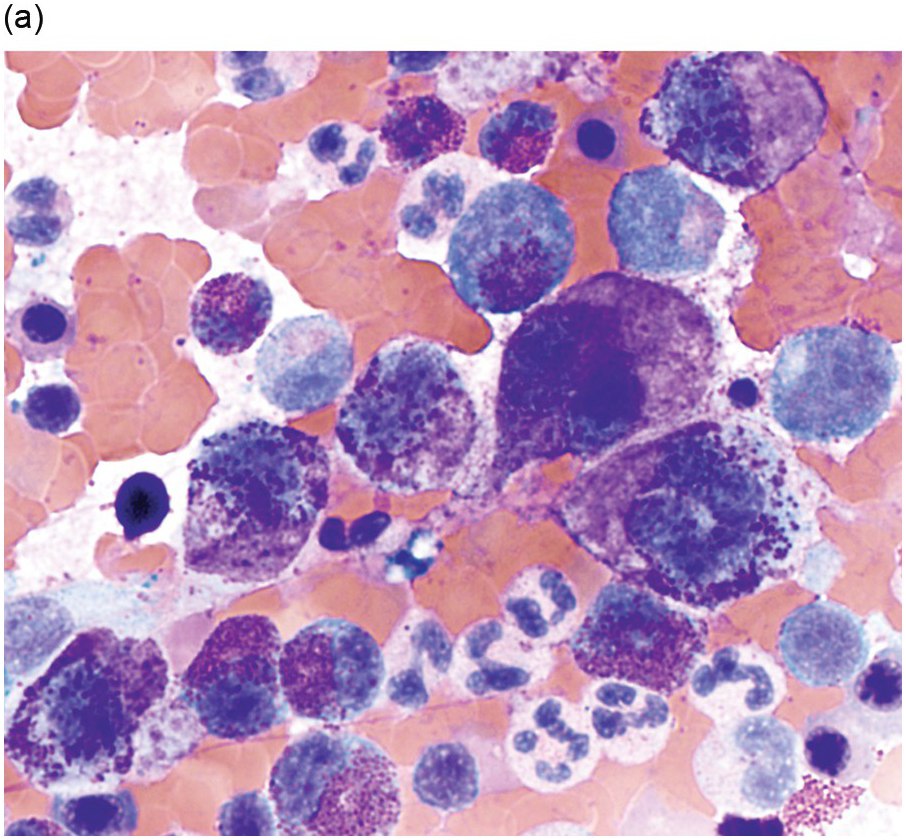
(a) Bone marrow aspirate smear showed large round immature mast cells with bilobed forms, multinucleated mast cells and irregular nuclei. Abundant cytoplasm with fine metachromatic granules was present and chromatin was partially condensed. (Bone marrow aspirate smear, Wright–Giemsa stain.)
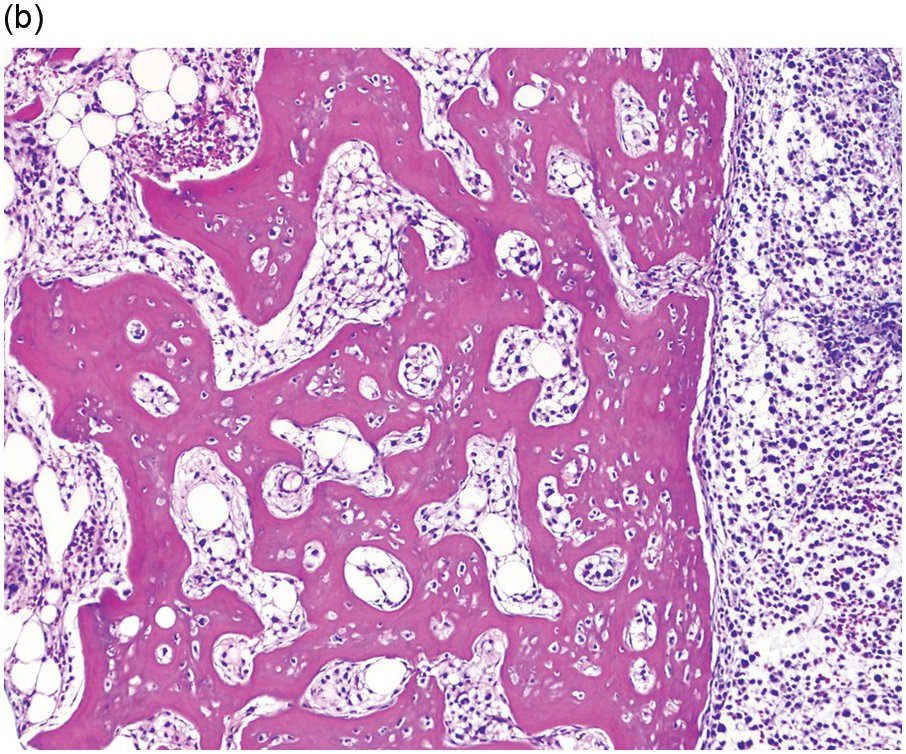
(b) A low power image from the bone marrow biopsy found marked osteosclerosis with sheets of neoplastic mast cells at right. (Bone marrow trephine, H&E.)
Figure 12.7 Mast cell leukaemia, aleukaemic variant. This 66-year-old female presented with ‘anaphylaxis’ and respiratory distress and was found to have eosinophilia. Imaging studies showed multiple sclerotic bone lesions. Peripheral blood smear showed mature eosinophils, but no circulating mast cells (not shown).
Subtypes
Classic leukaemic and aleukaemic variants of MCL are defined based on the number of circulating mast cells in the peripheral blood. Both subtypes still show greater than 20% mast cells on BM smears. The classic leukaemic variant shows circulating mast cells greater than 10% of total white blood cells in the peripheral blood, which differs from the aleukaemic variant with less than 10% circulating mast cells (Table 12.5). The aleukaemic variant of mast cell leukaemia is much more frequently encountered; however, it is not uncommon that patients may transition from the aleukaemic form to the leukaemic phase [9, 20].
Table 12.5 Variants of mast cell leukaemia.
| Mast cell leukaemia variants | Features/Criteria |
|---|---|
| Primary | No previous diagnosis of SM |
| Secondary | Transformation to MCL from a previous SM |
| Leukaemic | >10% circulating mast cells in peripheral blood |
| Aleukaemic | <10% circulating mast cells in peripheral blood |
| Acute | C-findings present, aggressive clinical coursea |
| Chronic | No C-findings, less aggressive clinical course |
a Can be with or without the presence of pathologic fractures; fractures due to osteoporosis do not qualify as a ‘C’ finding.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


