Cellular and Neurochemical Aspects of the Aging Human Brain: Introduction
Many cellular and molecular aspects of brain aging are shared with other organ systems, including increased oxidative damage to proteins, nucleic acids and membrane lipids, impaired energy metabolism, and the accumulation of intracellular and extracellular protein aggregates. However, as a result of the molecular and structural complexity of neural cells, which express approximately 50 to 100 times more genes than cells in other tissues, there are age-related changes that are unique to the nervous system. For example, complex cellular signal transduction pathways involving neurotransmitters, trophic factors, and cytokines that are involved in regulating neuronal excitability and plasticity are subject to modification by aging. This chapter describes cellular and molecular changes that occur in the brain during aging and how such changes may predispose neurons to degeneration in disorders such as Alzheimer’s disease (AD), Parkinson disease (PD), and Huntington disease (HD).
Structural Changes in the Aging Brain
All of the major cell types in the brain undergo structural changes during aging. These changes include nerve cell death, dendritic retraction and expansion, synapse loss and remodeling, and glial cell (astrocytes and microglia) reactivity. Such structural changes may result from alterations in cytoskeletal proteins and the deposition of insoluble proteins such as tau and α-synuclein inside of cells and amyloid in the extracellular space. Alterations in cellular signaling pathways that control cell growth and motility may contribute to both adaptive and pathological structural changes in the aging brain.
The cell cytoskeleton consists of polymers of different sizes and protein compositions. The three major types of polymers are actin microfilaments (6 nm in diameter); microtubules (25 nm in diameter), comprising of tubulin; and intermediate filaments (10–15 nm in diameter), which are made of specific intermediate filament proteins that are different in different cell types (e.g., neurofilament proteins in neurons and glial fibrillary acidic protein in astrocytes). To regulate the processes of filament assembly and depolymerization, and to link the cytoskeleton to membranes and other cell structures, neurons and glial cells employ an array of cytoskeleton-associated proteins. For example, neurons express several microtubule-associated proteins (MAPs) that are differentially distributed within the complex architecture of the cells; MAP-2 is present in dendrites but not in the axon, whereas tau is present in axons but not in dendrites. While there are no major changes in the levels of the most abundant cytoskeletal proteins with aging, there are changes in the cytoskeletal organization and in posttranslational modifications of cytoskeletal proteins. For example, increased amounts of phosphorylated tau occur in some brain regions, particularly those involved in learning and memory (e.g., hippocampus and basal forebrain). In addition, there is evidence that calcium-mediated proteolysis of MAP-2 and spectrin (a protein that links actin filaments to membranes) is increased in some neurons during aging. Oxidation of certain cytoskeletal proteins is suggested by studies demonstrating their modification by glycation and covalent binding of the lipid peroxidation product 4-hydroxynonenal (see “Free Radicals and the Aging Brain” later in this chapter). A consistent feature of brain aging in humans and laboratory animals is an increase in levels of glial fibrillary acidic protein, which may represent a reaction to subtle neurodegenerative changes.
Synapses are dynamic structural specializations where neurotransmission and other intercellular signaling events occur. There is considerable evidence for synaptic “remodeling” in the brain as we age, which is likely related to changes in dendritic arbors and neuronal numbers. For example, there may be decreases in synaptic numbers in some brain regions, but these may be offset by increases in the size of the remaining synapses. In other brain regions, no loss of synapses can be discerned.
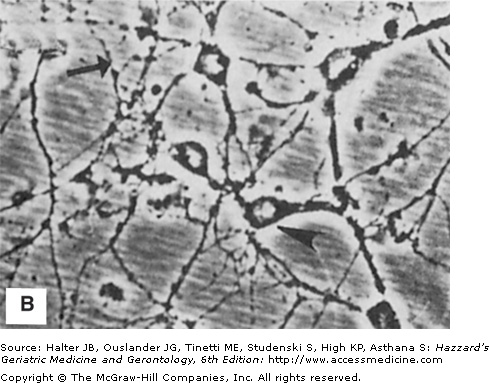
In contrast to usual aging, striking alterations in the neuronal cytoskeleton and synapses occur in neurodegenerative disorders such as AD, PD, and HD. Neurofibrillary tangles are filamentous accumulations of tau protein that form in the cytoplasm of degenerating neurons (Figure 61-1). Neurons with tangles have a decreased number of microtubules and often exhibit accumulations of MAP-2 and tau in the cell body. Tau is excessively phosphorylated in neurofibrillary tangles, which may result from reduced phosphatase activity as a consequence of oxidation or covalent modification by lipid peroxidation products. In PD, structures called Lewy bodies form in neurons and comprise abnormal accumulations of neurofilaments, with associated MAPs (particularly MAP-1b) and actin-related proteins such as gelsolin. In amyotrophic lateral sclerosis (ALS), lower motor neurons are filled with massive accumulations of neurofilaments that concentrate in proximal regions of the axon. While the specific molecular events involved in formation of the cytoskeletal alterations in different neurodegenerative disorders have not been established, there is increasing evidence for major roles of aberrant elevation of intracellular calcium levels and increased oxidative stress. Conditions that elevate intracellular calcium levels (e.g., exposure to glutamate or calcium ionophores) and induce oxidative stress (e.g., exposure to Fe2+ and amyloid β-peptide [Aβ], or expression of mutant Cu/ZnSOD) can elicit changes in the cytoskeleton in experimental animal and cell culture models that are similar to those seen in the human disorders (Figures 61-1 and 61-2).
Figure 61-1.
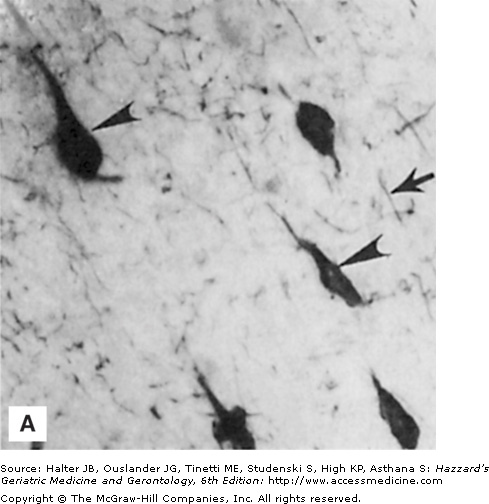
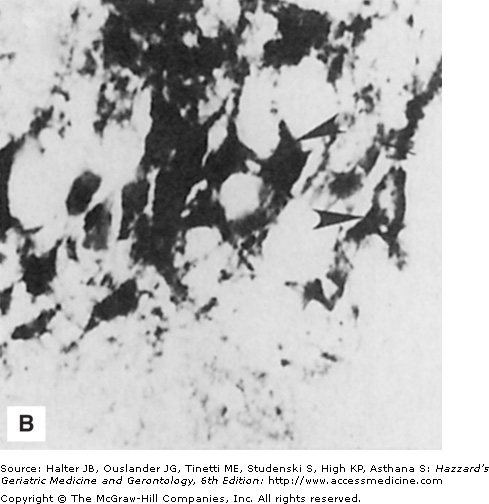
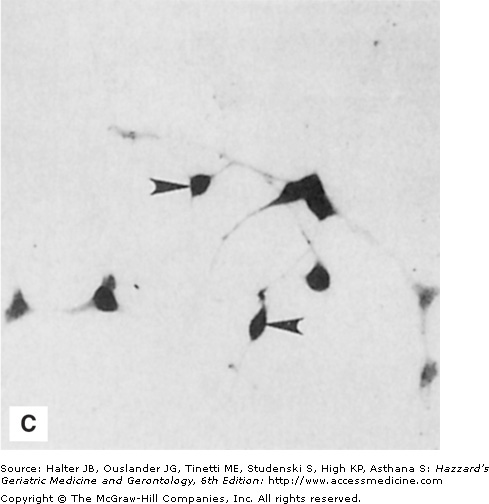
Neuronal cytoskeletal pathology in Alzheimer’s disease is mimicked in experimental systems by insults that increase intracellular calcium levels and induce oxidative stress. (A) Section of hippocampal tissue from an AD patient immunostained with an antibody against the microtubule-associated protein tau. Note strong staining of degenerating “tangle-bearing” neuronal cell bodies (arrowheads). (B) Section of hippocampus from an adult rat that had been administered the seizure-inducing excitotoxin kainate 6 hours previously. The section was immunostained with an antibody against the microtubule-associated protein tau. Note strong staining of degenerating neurons (arrowheads). (C) Human embryonic cerebral cortical neurons in culture that were left untreated (control) or were exposed for 2 hours to a calcium ionophore. Cells were then stained with an antibody against ubiquitin, a stress-responsive protein present in degenerating neurons in AD.
Figure 61-2.
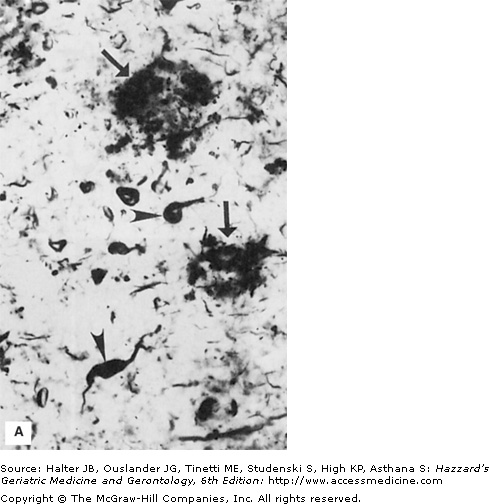
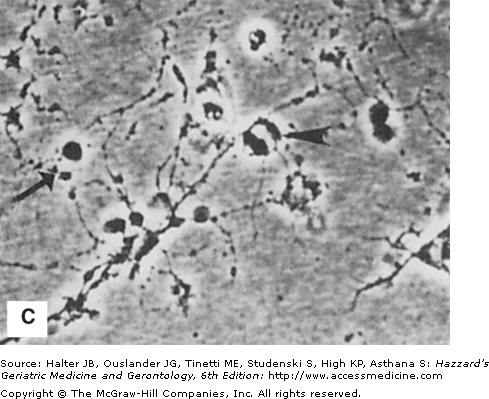
Histologic and experimental evidence that amyloid deposition plays a role in the neurodegenerative process in Alzheimer’s disease. (A) Section of hippocampal tissue from an AD patient immunostained with an antibody against the microtubule-associated protein tau. Note strong staining of degenerating “tangle-bearing” neuronal cell bodies (arrowheads) and neurites associated with amyloid plaques (arrow). (B and C) Phase-contrast micrographs of cultured rat hippocampal neurons that had been exposed for 24 hours to amyloid β-peptide (Aβ) alone (B) or in combination with corticosterone (C). Note that neurites in the culture exposed to Aβ alone are beginning to exhibit signs of damage (arrow), while the neuronal cell bodies still appear undamaged (arrowhead). Corticosterone exacerbated the damage to neurons exposed to Aβ. (See Goodman et al. J Neurochem. 1996;66:1836.)
Synapse loss occurs in neurodegenerative disorders and is strongly correlated with clinical symptoms. Accumulating data suggest that synaptic degeneration, resulting from excitotoxic events localized to synapses, may initiate the neuronal death process in AD, PD, HD, and stroke. Glutamate receptors are highly concentrated in postsynaptic dendritic spines, which represent sites of massive calcium influx during normal physiologic synaptic transmission. Age-related decreases in energy availability and increases in oxidative stress, and disease-specific alterations such as Aβ accumulation in AD and trinucleotide expansions of the huntingtin protein, may render synapses vulnerable to excitotoxic injury (see below).
As in other organ systems, vessels that supply blood to the brain are vulnerable to age-related atherosclerosis and arteriosclerosis, which render the vessels susceptible to occlusion or rupture (stroke), a major cause of disability and death in the elderly population. Reduced brain perfusion in the absence of overt stroke may play a role in age-related cognitive dysfunction. Decreased cerebral blood flow occurs with advancing age and is accompanied by declines in cerebral metabolic rate for oxygen and glucose use. Age-related changes in cerebral vasculature are generally similar to those that occur in vessels elsewhere in the body and are therefore likely to result from common cellular and molecular changes, including oxidative damage to vascular endothelial cells and an inflammatory response in which macrophages may penetrate the blood–brain barrier (Figure 61-3).
Figure 61-3.
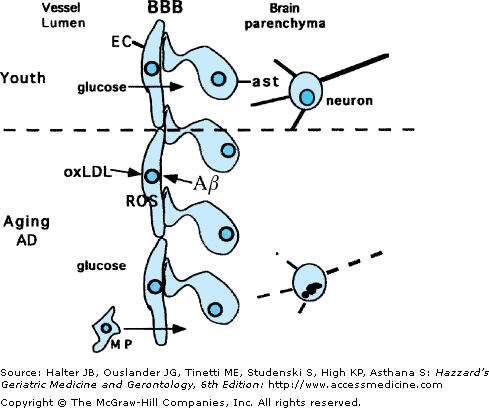
Adverse effects of aging and Alzheimer’s disease on endothelial cells and the blood–brain barrier. Vascular endothelial cells (EC) and associated astrocyte (ast) processes form the blood–brain barrier (BBB), which plays important roles in transporting nutrients (e.g., glucose) into the brain, while at the same time preventing movement of toxic substances and blood cells such as macrophages (MP). With aging, and more so in AD, oxidative damage to endothelial cells results from exposure to oxidized LDL and amyloid β-peptide (Aβ). Oxidative stress impairs transport functions of the EC and promotes penetration of MP into the brain parenchyma. Such compromise of the vasculature results in reduced nutrient availability to neurons and inflammatory processes that promote degeneration of neurons.
Age-dependent cerebral vascular changes are strongly linked to heart disease and hypertension. Interestingly, apolipoprotein E polymorphisms are linked to increased risk of both atherosclerosis and AD, with the apolipoprotein E4 increasing the risk. This association suggests that age-related vascular changes may make an important contribution to the neurodegenerative process in AD. Finally, important transport functions of cells (endothelial cells and astrocytes) that comprise the blood–brain barrier may also be impaired in the aging brain and more so in AD. Many of the same behavioral and dietary approaches now recognized to forestall cardiovascular disease may also forestall cerebrovascular disease; these include engaging in physical and mental activities, a low calorie intake, and a high antioxidant intake.
Aβ is a 40- to 42-amino-acid peptide that arises from a much larger membrane-spanning β-amyloid precursor protein (APP). During normal aging, and to a much greater extent in AD, Aβ forms insoluble aggregates (plaques) in the brain parenchyma and vasculature (Figure 61-4).
Figure 61-4.
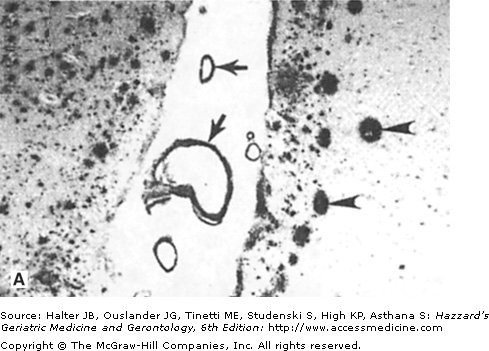
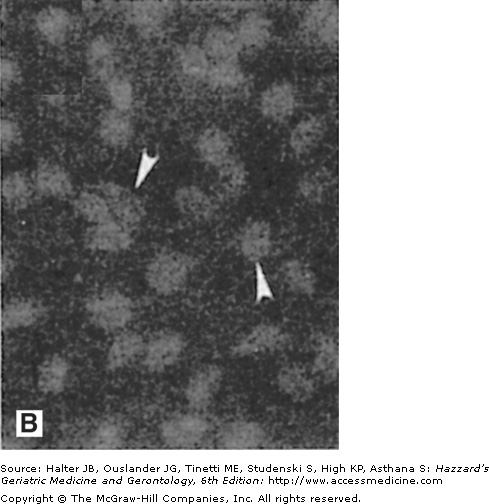
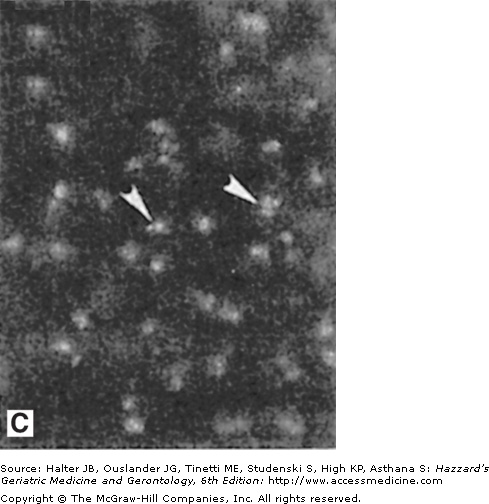
Amyloid deposits in the brain parenchyma and cerebral vessels: evidence that amyloid damages vascular endothelial cells. (A) Section of hippocampal tissue from an AD patient. The section was immunostained with an antibody against amyloid β-peptide (Aβ). Note numerous Aβ deposits of various sizes in the brain parenchyma (arrowheads point to large plaques) and cerebral blood vessels (arrows). (B and C) Cultured vascular endothelial cells were exposed for 24 hours to saline (B) or Aβ (C), and were then stained with the fluorescent deoxyribonucleic acid (DNA)-binding dye HOECHST 33342. The nuclear DNA in cells not exposed to Aβ exhibits the normal diffuse uniform distribution, while the chromatin in the cells exposed to Aβ exhibits condensation and fragmentation consistent with apoptotic death. (Adapted from Blanc EM, Toborek M, Mark RJ, et al. Amyloid β-peptide disrupts barrier and transport functions and induces apoptosis in vascular endothelial cells. J Neurochem. 1997;68:1870.)
Amyloid plaques accumulate most heavily in brain regions involved in learning and memory processes such as the hippocampus and entorhinal cortex. In AD, the accumulation of Aβ in the brain is correlated with the amount of neuronal degeneration and with the severity of cognitive impairment. Although the cause(s) of the majority of cases of AD is unknown, some AD cases are caused by mutations in APP, presenilin-1, or presenilin-2; mutations in each of these proteins result in increased production of self-aggregating neurotoxic forms of Aβ. Aβ can be neurotoxic, and can increase neuronal vulnerability to metabolic, excitotoxic, and oxidative insults. The sequence of events involved in Aβ-induced neuronal injury and death may involve induction of membrane lipid peroxidation, which generates a toxic aldehyde called 4-hydroxynonenal that covalently modifies proteins involved in maintenance of cellular ion homeostasis and energy metabolism, including the plasma membrane Na+/K+ and Ca2+ ATPases (adenosine triphosphatases), glucose transporters, and glutamate transporters. This may lead to excessive elevation of intracellular calcium levels, mitochondrial dysfunction, and a form of cell death called apoptosis. In addition to its likely involvement in the neurodegenerative process, Aβ may also disrupt neurotransmitter signaling pathways. For example, Aβ impairs coupling of muscarinic acetylcholine receptors to their downstream GTP (guanosine triphosphate)-binding effector protein, an action that likely contributes to the well-established deficits in cholinergic signaling pathways in AD. Finally, Aβ may also contribute to vascular damage and inflammatory processes in the aging brain.
Free Radicals and the Aging Brain
A free radical is any molecule with an unpaired electron in its outer orbital; in biological systems, oxygen-based molecules are the predominant free radical species (Figure 61-5). A major oxyradical in cells O2−. is superoxide (O2−.), which is generated during the mitochondrial electron transport process; superoxide dismutases (MnSOD and Cu/ZnSOD) eliminate O2−. by converting it to hydrogen peroxide (H2O2). H2O2 can be converted to hydroxyl radical (OH•) via the Fenton reaction, which is catalyzed by Fe2+ and Cu+. Peroxynitrite (ONOO−) arises from the interaction of nitric oxide (NO) with O2−.; calcium influx is a major stimulus for NO production. While OH• and ONOO− can cause direct damage to proteins and deoxyribonucleic acid (DNA), their major means of damaging cells is by attacking fatty acids in membranes and initiating a process called lipid peroxidation. Studies of aging have provided compelling evidence that there is an increase in production and accumulation of oxyradicals in essentially all tissues in the body during the aging process, including in the brain.
Figure 61-5.
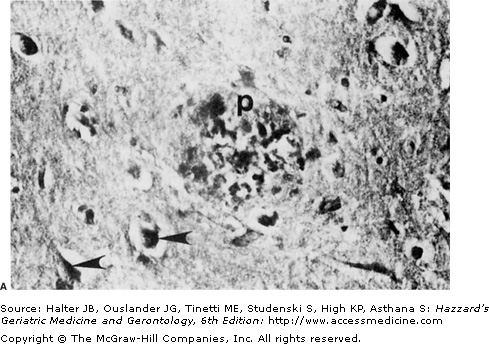
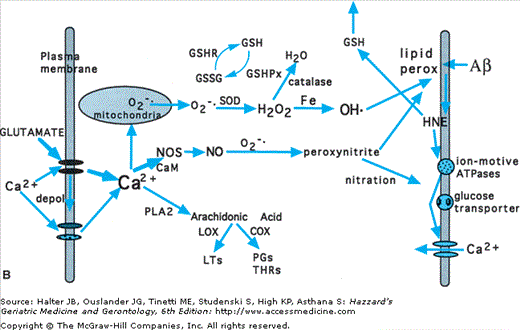
Possible sources of oxidative stress contributing to dysfunction and degeneration of neurons in aging and Alzheimer’s disease. (A) Section of hippocampal tissue from an AD patient. The section was immunostained with an antibody that recognizes lipid peroxides that are by-products of membrane lipid peroxidation. Note immunoreactivity associated with neuritic plaques (p) and neurofibrillary tangles (arrowheads). (B) The major cellular source of reactive oxygen species in neurons is mitochondria where O2−. is generated during the electron transport process. The superoxide dismutase enzymes (SOD) convert O2−. to H2O2. Fe2+ catalyzes the production of OH• from H2O2, while glutathione peroxidase (GSHPx) and catalase detoxify H2O2. Peroxynitrite can be formed by the interaction of O2−. with nitric oxide (NO). Both OH•, and peroxynitrite induce membrane lipid peroxidation (MLP), which may occur in the plasma membrane, mitochondrial membranes, and endoplasmic reticulum (ER) membranes. During aging and in age-related neurodegenerative disorders, agents such as amyloid β-peptide (Aβ) can induce MLP. MLP liberates the aldehyde 4-hydroxynonenal (HNE), which impairs the function of membrane transporters (glucose and glutamate transporters, and ion-motive ATPases) and ion channels, and thereby alters their activities. Reduced glutathione (GSH) binds and detoxifies HNE, thereby serving an important neuroprotective role. The antiapoptotic gene product Bcl-2 may act, in part, by suppressing MLP in plasma, mitochondrial, and ER membranes. Elevation of intracellular calcium levels, as induced by glutamate, promotes oxyradical production and MLP by inducing NO and O2−. production, and by activating phospholipases resulting in production of arachidonic acid, which is then acted upon by cyclooxygenases (COX) and lipoxygenases (LOX). (Modified from Mattson MP. Trends Neurosci. 1997;20:395.)
Levels of lipid peroxidation products such as lipid peroxides and 4-hydroxynonenal are significantly elevated in the brain parenchyma and cerebrospinal fluid in AD (see Figure 61-5). Immunohistochemical analyses of brain tissue from AD and PD patients, and spinal cord tissue from ALS patients, using antibodies directed against 4-hydroxynonenal adducts revealed increased levels of 4-hydroxynonenal in association with degenerating neurons and neuritic plaques suggesting a role for this aldehyde in the neurodegenerative process. Studies of cell culture and animal models of AD and ALS show that lipid peroxidation can impair the function of the plasma membrane glucose and glutamate transporters and renders neurons vulnerable to excitotoxicity and apoptosis. Administration of antioxidants such as vitamin E to cultured cells and transgenic mice expressing mutations that cause AD or ALS can retard the neurodegenerative process, suggesting a causal role for lipid peroxidation in these diseases.
Levels of protein oxidation, measured as protein carbonyls, are significantly elevated in the brains of aged rodents, and such age-associated protein oxidation can be prevented by administration of antioxidants. Studies of membrane structure have provided evidence for oxidation-related alterations in membrane protein conformation in old rodents. Studies of brain tissues of patients with AD and PD have revealed increased levels of protein oxidation in vulnerable brain regions and, in particular, in degenerating neurons. Oxidation of proteins impairs their function and is therefore a likely contributor to age-related cellular dysfunction and neuronal degeneration. Progressive addition of sugar residues to many different proteins occurs during the aging process. This process, called glycation, may promote oxidative stress in cells. Two proteins that have been shown to be heavily glycated in AD are Aβ and tau, the major components of plaques and neurofibrillary tangles, respectively.
Very little nuclear DNA damage occurs in the nervous system during usual aging, but DNA damage may contribute to the pathogenesis of neurodegenerative disorders. Damage to nuclear DNA in striatum of HD patients, and in hippocampus and vulnerable cortical regions of AD patients, has been documented. Specifically, levels of 8-hydroxyguanosine are increased. This DNA damage may be caused by oxyradicals, with hydroxyl radical and peroxynitrite being the major culprits. Progressive oxidative damage to mitochondrial DNA occurs during aging, and may be exacerbated in neurodegenerative disorders. Mitochondrial DNA is particularly prone to damage because this organelle is the site where the vast majority of free radicals are generated and because cells do not possess effective systems for repair of damaged mitochondrial DNA. Damage to mitochondrial DNA can lead to failure of electron transport and reduced adenosine triphosphate (ATP) production. Moreover, the important calcium sequestering function of mitochondria may be compromised as the result of age-related DNA damage, which may increase neuronal vulnerability to excitotoxicity and apoptosis.