Cell-Mediated Cytotoxicity
Judy Lieberman
INTRODUCTION
The most effective way the immune system can control the threats of intracellular infection and cellular transformation is by destroying infected and cancerous cells.1,2,3,4,5,6,7 When killer lymphocytes recognize harmful cells, they can target them for elimination by triggering programmed cell death. The main killer cells are natural killer (NK) cells of the innate immune sytem and cluster of differentiation (CD)8+ T lymphocytes of adaptive immunity, although some CD4+ T lymphocytes, particularly TH1 and regulatory T (Treg) cells, also express and deploy the specialized cell death machinery. All killer lymphocytes contain specialized secretory lysosomes, called cytotoxic granules, that are filled with deathinducing enzymes, called granzymes (“granule enzyme”). When the killer cell is activated, the cytotoxic granules move to the immune synapse formed with the target and fuse their membranes with the killer cell membrane, dumping their contents into the immune synapse in a process termed granule exocytosis. Perforin, a pore-forming protein in the granules, delivers the death-inducing granyzmes into the cytoplasm of the target cell to initiate its death. In this encounter, the killer cell remains unharmed.8 It is a serial killer that can detach from one target to seek and destroy others.9 Killer cells can also activate programmed cell death by using cell surface receptors to ligate cellular death receptors, such as Fas, on target cells. Granule-mediated cell death is key to control viral and intracellular bacterial infection and cancer because perforin-deficient mice and humans homozygous for perforin mutations or deficient in molecules needed for granule exocytosis are highly vulnerable to infection with intracellular pathogens and prone to develop spontaneous lymphomas.10 The death receptor pathway regulates lymphocyte homeostasis. Patients genetically deficient in the death receptor Fas or its ligand FasL develop autoimmunity.11 Target cells destroyed by cytotoxic granules or death receptor ligation die a highly regulated death (programmed cell death or apoptosis) rather than by necrosis. Programmed cell death minimizes inflammation and damage to nearby tissue as target cells undergoing programmed cell death are rapidly recognized and cleared by immune phagocytes, especially macrophages.12 The topic of this chapter was reviewed in more depth in a recent issue of Immunological Reviews.13
In this chapter we first describe the killer cells: Which immune cells are able to kill and how they develop this capacity and are regulated. Because of its destructive potential, cytotoxicity needs to be carefully regulated. We next focus on the death machinery used for granule- and death receptor-mediated cytotoxicity and how it is mobilized and used to destroy the target cell. We also discuss what is known about how killer cells are protected against their own weapons of destruction. Some granzymes are expressed without perforin in nonkiller cells. We also discuss the increasing evidence for noncytotoxic proinflammatory roles of killer molecules.
THE KILLER CELLS
The major killer cells are NK cells in innate immunity and CD8 T cells in adaptive immunity. Naïve T cells that have not previously seen antigen do not express either granule effector molecules or death receptors and are incapable of cell-mediated cytotoxicity.14 Within about 5 days of activation, naïve CD8 T cells differentiate into effector cytotoxic T lymphocytes (CTLs) that express both types of cytotoxic molecules. At the same time, these cells downregulate adhesive and chemokine receptor molecules that retain them in lymph nodes and acquire receptors that allow them to traffic to tissue sites of infection and tumor invasion. Activation to cytotoxic effector cells is tightly regulated. It requires not only antigen-receptor activation but also costimulation, and is greatly enhanced when antigen-presenting cells are stimulated by danger and pathogen-associated pattern motif receptors or when naïve T cells are stimulated by exogenous inflammatory and antiviral cytokines, including type I interferons (IFNs), interleukin (IL)-1, and IFN γ. Upon activation, effector CD8 T cells also begin to express the Fcγ receptor CD16, also present on NK cells, which enables them to recognize and lyse target cells that have been coated with IgG antibodies in a process called antibody-dependent cell-mediated cytotoxicity.15 In situations of persistent and extensive antigen, however, such as occur in tumors and chronic viral infection, many of the CD8 T cells that have the surface protein expression of CD8 effector cells no longer express perforin and are not cytotoxic.16,17,18 Effector CD8 T cells that lack cytotoxicity have been termed “exhausted.” Most effector cells in an immediate immune response die within a few weeks, but some survive and develop into memory cells. Memory cells downregulate expression of cytotoxic effector proteins, but the kinetics of downregulation varies with the molecule and with the particularities of the immunostimulatory environment.14,19 In particular, activation of CD8 T cells without CD4 T-cell help leads to an unimpaired primary cytotoxic response but greatly impairs the development of antigen-specific memory cells.20
The immunosuppressive drug rapamycin directs antigenstimulated CD8 T cells to differentiate preferentially into memory cells rather than to effector CTLs.21 Memory CD8 T cells rapidly reacquire cytotoxic capability within hours of restimulation. The molecular basis for this rapid response is not well understood, although recent studies suggest that in memory CD8 T cells, the chromatin of cytolytic effector gene promoters and of eomesodermin, the master transcription factor that regulates CD8 effector genes, bears epigenetic marks that poise them for transcription compared to naïve T cells.22,23,24,25,26 These cells may also store messenger ribonucleic acids (mRNAs) for perforin and granzyme that can be rapidly translated upon activation. Some types of activated CD4 T cells, especially TH1, NKT, and Treg cells, also express granzymes and perforin and have cytotoxic activity. Murine Tregs express granzyme B but probably not granzyme A.27,28 Although immunosuppression by Tregs is mediated by soluble factors, there is also a poorly understoood component that requires cell-to-cell contact. Direct lysis of cognate T cells and potentially other immune cells by granule-mediated and death receptor pathways by Tregs is likely an important mechanism for suppressing immune activation.29,30,31
The immunosuppressive drug rapamycin directs antigenstimulated CD8 T cells to differentiate preferentially into memory cells rather than to effector CTLs.21 Memory CD8 T cells rapidly reacquire cytotoxic capability within hours of restimulation. The molecular basis for this rapid response is not well understood, although recent studies suggest that in memory CD8 T cells, the chromatin of cytolytic effector gene promoters and of eomesodermin, the master transcription factor that regulates CD8 effector genes, bears epigenetic marks that poise them for transcription compared to naïve T cells.22,23,24,25,26 These cells may also store messenger ribonucleic acids (mRNAs) for perforin and granzyme that can be rapidly translated upon activation. Some types of activated CD4 T cells, especially TH1, NKT, and Treg cells, also express granzymes and perforin and have cytotoxic activity. Murine Tregs express granzyme B but probably not granzyme A.27,28 Although immunosuppression by Tregs is mediated by soluble factors, there is also a poorly understoood component that requires cell-to-cell contact. Direct lysis of cognate T cells and potentially other immune cells by granule-mediated and death receptor pathways by Tregs is likely an important mechanism for suppressing immune activation.29,30,31
Because it takes a week to 10 days for naïve CD8 T cells to proliferate and differentiate into a large population of antigen-specific CTLs, the immediate response to intracellular infection in individuals that have not been vaccinated or previously exposed is mediated by NK cells. Although freshly minted NK cells were previously thought to immediately express granzymes and perforin, it now seems clear that—at least in mice—resting NK cells have minimal cytotoxic activity.32 They constitutively express mRNAs for granzymes A and B and perforin but only have granzyme A protein. Because they lack substantial perforin protein, cytotoxicity is limited. However, perforin and granzyme proteins and cytotoxicity are upregulated rapidly when NK cell-activating receptors are stimulated. Less differentiated NK cells that highly express the neural cell adhesion molecule or CD56 are poorly cytotoxic, whereas more differentiated CD56dim NK cells are potent killer cells.33 In the circulation, CD56dim NK cells have about a log more perforin than CD56bright NK cells. NK-activating receptors recognize cell surface changes in tumors, stressed cells, and infected cells, such as downregulation of major histocompatibility complex/human leukocyte antigen molecules or cell surface expression of nonclassical major histocompatibility complex molecules, such as MICA and MICB, which are induced by stress. A longstanding dogma of innate immunity is that innate immune responses are not altered by antigen exposure. However, it is now clear that NK cytotoxicity to infection and other stimuli can be greatly increased by previous antigen exposure.34 NK cell memory of prior exposure probably results from the expansion of NK cells bearing activating receptors specific for different important pathogens. These receptors, many of which are poorly conserved during mammalian evolution, may have coevolved with important pathogens. The link between individual NK receptors and pathogen recognition remains to be defined.
CYTOTOXIC GENE EXPRESSION
There are 5 human granzymes and 10 mouse granzymes expressed from three gene clusters that arose by gene duplication. In humans, the genes encoding granzymes A and K, tryptases that cleave after basic amino acids, are clustered on chromosome 5; the genes for granzyme B, which cleaves after aspartic acid residues such as the caspases; and granzyme H (or C in mice), which cleaves after hydrophobic residues, are clustered with myeloid cell proteases like mast cell chymase on chromosome 14; and the gene for granzyme M, which is highly expressed in NK cells and cleaves after Met or Leu, is found on chromosome 19 (Fig. 37.1). The mouse granzyme B cluster is uniquely expanded by multiple gene duplications to encode, in addition, granzymes D, E, F, G, L, and N. Nothing is known about these mouse-specific enzymes, but they may have evolved to defend against specific common mouse pathogens.1 Granzyme A and B are the most abundant granzymes and the most studied. Killer cells, including NK cells, cytotoxic CD4 and CD8 T cells, and even some Treg cells, express highly individualized and tightly regulated patterns of granzymes that depend on both cell type and mode of activation.35,36
Expression in Noncytolytic Cells
Perforin is only expressed by cytotoxic cells. Although granzymes were previously also thought to have similarly restricted expression, noncytotoxic cells can express granzymes without perforin.37 Granzyme transcripts can be amplified from prothymocytes in fetal liver and double negative thymocytes.38 Although granzyme A transcripts are detected in thymocytes with the potential to develop into CD8+ cells, granzyme A activity is detected only in the most mature CD4-CD8+ thymocytes. These results suggest posttranscriptional regulation of granzyme translation (see the following for additional examples). Granzyme B, but not granzyme A, is expressed in Treg cells and plays an important perforin-dependent role in Treg function in mice. Benign and transformed B cells can be induced to express granzyme B by IL-21 alone or when combined with anti-B-cell receptor antibody.39 Granzyme B is also expressed without perforin in many different types of myeloid cells. Within the immune system, granzyme B is expressed in human plasmacytoid dendritic cells (pDCs).40 There are comparable levels of granzyme B transcripts in resting and activated pDCs but significantly higher amounts of granzyme B protein in activated cells, suggesting posttranscriptional regulation of expression. Granzyme B is also expressed in both normal and neoplastic human mast cells in vitro and in vivo.41 It localizes to mast cell granules and is secreted when they are activated. In mice, skin-associated mast cells and bone marrow-derived in vitro differentiated mast cells express granzyme B but lung mast cells do not.42 Neither granzyme A nor perforin are detected in mouse mast cells. The granzyme B gene is encoded within a few hundred kilobases of mast cell proteases. Thus, the granzyme B/mast cell chymase and tryptase genomic region is likely open and active in mast cells. In human basophils, IL-3 induces granzyme B, but not granzyme A or perforin,
expression.43 Expression of granzyme B in mast cells and basophils suggests a role of granzyme B in mediating allergic disease. In fact, granzyme B has been found in bronchoalveolar lavage fluid after allergen exposure. Several studies have suggested that granzyme B and perforin are expressed in human neutrophils, but this is controversial.44,45,46,47
expression.43 Expression of granzyme B in mast cells and basophils suggests a role of granzyme B in mediating allergic disease. In fact, granzyme B has been found in bronchoalveolar lavage fluid after allergen exposure. Several studies have suggested that granzyme B and perforin are expressed in human neutrophils, but this is controversial.44,45,46,47
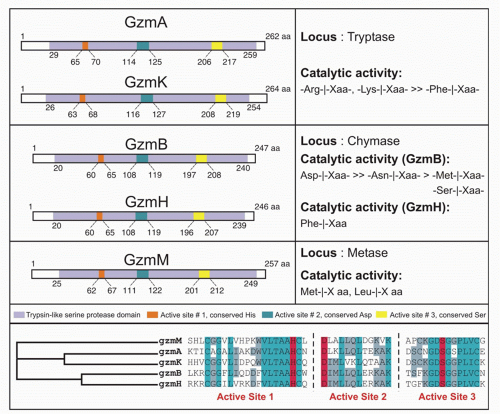 FIG. 37.1. The Human Granzymes are Encoded in Three Clusters. (Figure reprinted with permission from Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26: 389-420.) |
Granzyme B is also expressed in the absence of perforin in the human reproductive system in developing spermatocytes and in placental trophoblasts48 and by granulosa cells of the human ovary in response to follicle stimulating hormone.49 In addition, granzyme B has been detected in a subset of primary human breast carcinomas and in chondrocytes of articular cartilage.50 The granzyme M transcript is expressed at low levels in the photoreceptor cells of the retina in the mouse.51 An alternatively spliced form (aGM) is exclusively expressed in these cells at much higher levels. Like granzyme M, granzyme K has an alternatively spliced form exclusively expressed in the brain.52 The physiologic significance of the alternative transcripts of granzymes M and K is unclear.
Extracellular Signals Regulating Granzyme Expression
The kinetics and expression of the individual granzymes and perforin vary in different clonal populations in vitro and in vivo and depend on how they are activated.53,54,55 Most circulating CD8+ T lymphocytes that express any granzyme express both granzyme A and granzyme B, but some cells are positive for only one granzyme. Single-cell expression profiles of granzymes, perforin, and IFNγ have been investigated in in vitro or in vivo activated CD8+ T cells using reverse transcription-polymerase chain reaction in mice35 and intracellular staining and flow cytometry in humans.56 Individual T cells show diverse expression of these genes. Although some pairs of genes (perforin and IFNγ) are coexpressed more frequently than others, no specific combination of genes is consistently coexpressed. During in vitro activation of mouse naïve lymphocytes with antibodies to CD3, CD8, and CD11a and IL-2, the expression of granzyme A and granzyme C is delayed compared with cytolytic activity and expression of perforin and granzyme B.35 When mouse CTLs are activated in vivo by influenza virus infection, most antigen-specific CD8 T cells found in the lung 1 week after infection express both granzymes A and B, and about a third of them also express perforin. Moreover, there is no in vivo difference in the kinetics of induction of granzyme A, granzyme B, or perforin. Granzyme C is not induced by influenza infection in vivo. The diversity of expression of individual granzyme and perforin genes suggests that each gene is regulated independently, although it is likely that these genes will share some common transcription factor recognition sites and epigenetic changes. Differences in T-cell receptor (TcR) avidity, costimulatory and inhibitory receptor engagement, danger and innate immune receptor activation, cytokine milieu, type and state of activation of the antigen-presenting cell, and presence of helper or regulatory CD4 T cells will likely influence the induction of the granzyme and perforin genes. Moreover, the cell’s prior history of activation will affect cytolytic gene expression during subsequent encounters with antigen. Surprisingly, little is known about this subject.
The perforin and granzyme genes are induced during T-cell activation. However, the only signal shown consistently to upregulate granzyme A and B and perforin is IL-2.57 IL-2 regulates perforin and granzyme expression directly and independently of its effect on CD8+ T-cell survival and proliferation.58 Mice genetically deficient in IL-2 retain the ability to elicit a CTL response against many viruses, tumors, and allografts,59,60 although there are deficiencies in cytotoxicity under certain conditions.61 The other γc-dependent cytokines (IL-4, IL-7, IL-9, IL-15, and IL-21) likely substitute for IL-2 in its absence. IL-15 is particularly important because it also shares the γ-chain with the IL-2 receptor. IL-15 induces the expression of perforin, granzymes A and B, IFNγ, and Fas ligand in primary mouse lymphocytes.62 IL-21 works synergistically with IL-15 to upregulate granzyme A and B expression in mouse CD8 T cells.63 In vivo in mice, IL-21 exhibits potent antitumor function by enhancing NK and CD8 T-cell cytotoxicity.64 Similarly in human peripheral blood CD8 T cells, IL-15 and IL-21 both activate granzyme B and perforin expression, but IL-21 does so without inducing CD8 T-cell proliferation.65 Members of the IL-6/IL-12/IL-27 family also can upregulate granzyme and perforin expression.66,67
Transcriptional Regulation of Perforin and Granzymes
Two key transcription factors, T-bet (TBX21) and eomesodermin (EOMES), which belong to the T-box family, are the key master regulators of cytotoxic gene expression and survival of committed CD8 memory cells.68,69,70,71 After naïve CD8 T-cell activation, T-bet is induced before eomesodermin.72 Notch signaling and the Runx3 transcription factor upregulate eomesodermin, but also directly upregulate expression of perforin and granzyme B genes.72,73 Mice deficient in both T-bet and eomesodermin genes are unable to control tumors and intracellular infection.74,75,76 They develop a wasting syndrome caused by anomalous differentiation to IL-17-secreting cells, suggesting that these two genes not only positively regulate cytotoxic gene expression and other genes required for CTL survival and function but also suppress differentiation to alternate lineages.
Chromosome transfer experiments have shown that expression of the perforin gene (PRF1/prf1) is regulated by cis-regulatory regions extending about 150 kb around the gene.72,77,78 These include a core promoter located 120 bp upstream of the transcription start site and two enhancer regions and a locus control region (LCR) that are altered during T-cell differentiation and activation. The LCR is open for transcription specifically in cytotoxic cells. The region around the presumed LCR is more accessible to deoribonuclease (DNase I) digestion (and therefore its chromatin is open) in murine CD8 CTLs than in CD4 TH1 cells, likely explaining their approximately 20-fold increase in prf1 mRNA. Increased IL-2 does not enhance the accessibility of the LCR. The enhancers are both activated by IL-2R signaling mediated by signal transducer and activator of transcription (STAT)5 binding to two sites in each enhancer. Other STAT family members activated by alternate cytokines can also activate them. Activation of the more proximal enhancer also depends on IL-2-activated NF-κB binding. Both enhancers also contain binding sites for AP-1 and ETS transcription factors, whereas the distal enhancer has an E-box and NFAT binding site, and the proximal enhancer contains eomesodermin, Ikaros, and CREB binding sites. Recruitment of ribonucleic acid (RNA) pol II to the transcription start site and activation of transcription increase with IL-2 stimulation. The key factors involved in activating transcription at the prf1 promoter are Runx3 and eomesodermin. T-bet does not appear to play a direct role in activating prf1 transcription but likely acts indirectly by increasing IL2-Rβ expression and enhancing IL-2 signaling. The current model suggests that Runx3 is needed to open the extended prf1 locus during T-cell differentiation, whereas eomesodermin plays a more direct role in activating transcription near the promoter. Other transcription factors also likely participate in transactivating the perforin promoter, including an ETS transcription factor, probably myeloid elf-1-like factor (MEF).
Much less is known about the details of gene regulation of the granzymes. Enhancers or other long-range regulatory region of granzyme genes remain to be defined. Granzyme B is the only granzyme whose expression has been studied. A distal DNase hypersensitivity site 3.9 kb upstream of the granzyme B transcription start site is accessible only in activated, but not resting, CD8 T cells.79 Inclusion of this region in a GFP reporter in transgenic mice enhances CTL-specific expression, suggesting that this region may have enhancer activity. Induction of the expression of granzyme transcripts requires at least two independent stimuli: activation of the TcR and costimulation by cytokines of the γc family. The signals from several distinct signal transduction pathways are integrated in the nucleus in the form of transcription factors that bind to granzyme gene regulatory elements and activate transcription. Early studies identified a 243-bp fragment upstream of the mouse granzyme B transcription start site that potentially regulates granzyme B transcription.80 This region contains binding sites for two ubiquitous transcription factors: activating transcription factor/cyclic AMP-responsive element binding protein and activator protein-1, and two lymphoid specific factors, Ikaros and core-binding factor (PEBP2).81 Several of these transcription factor binding sites are evolutionarily conserved between the human and mouse granzyme B promoters.82,83 Analysis of reporter assays using promoters that had been systematically mutated at these sites in primary cells and cell lines revealed subtle differences in the importance of some transcription factors in primary cells versus cell lines. For example, activator protein-1, cyclic AMP-responsive element binding protein, and core-binding factor were not as important for transcription in primary cells as they appeared to be in cell lines.82,84 These studies suggested that combinations of transcription factors (particularly, activator protein-1 and corebinding factor) activate granzyme B expression in primary cells. The most compelling difference between the mouse and human granzyme B gene promoter is the importance of the Ikaros site only in human granzyme B expression.82,84 Studies in Stat1-deficient mice indicate that STAT1 mediates
granzyme B induction by IFNα or IL-27.67,85 IL-27-induced augmentation of granzyme B expression also depends on T-bet.67 Eomesodermin also drives granzyme B expression.71 Direct binding of T-bet and eomesodermin to the granzyme B promoter has not been examined.
granzyme B induction by IFNα or IL-27.67,85 IL-27-induced augmentation of granzyme B expression also depends on T-bet.67 Eomesodermin also drives granzyme B expression.71 Direct binding of T-bet and eomesodermin to the granzyme B promoter has not been examined.
Posttranscriptional Regulation
Several examples of cells expressing perforin and/or granzyme transcripts, but not protein, were described previously, including resting NK cells, thymocytes, and unactivated pDCs and mast cells. Murine memory CTLs also express abundant granzyme B mRNA but no protein.86 All these results point toward a general mechanism of “prearming” cytotoxic lymphocytes with effector mRNAs, allowing these cells to rapidly respond to external stimuli. This type of gene regulation is well known to regulate cytokine expression, presumably for the same purpose. Two recent studies provide evidence for negative regulation of granzyme B and perforin expression by microRNAs miR-27* and miR-223 in NK cells.87,88 It will be interesting to see if expression or processing of these micro RNAs declines rapidly after NK cell activation.
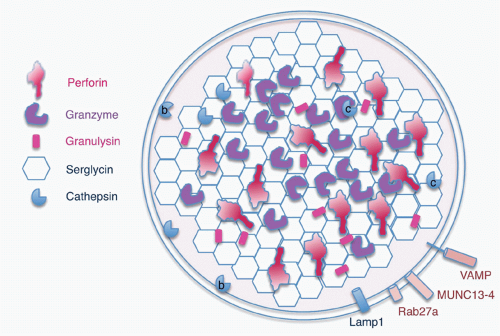 FIG. 37.2. Key Components of Cytotoxic Granules. The cytolytic effector molecules, perforin, granzymes, and granulysin, are bound to the serglycin proteoglycan. Cytotoxic granules also contain molecules found in all lysosomes, such as Lamp1 (CD107a) and cathepsins, as well as membrane-associated proteins specific to secretory lysosomes, such as vesicle-associated soluble N-ethylmaleimide-sensitive factor accessory complex component (VAMP)7 or VAMP8, Munc13-4, and Rab27a, which are essential for granule exocytosis. Cathepsins B and C play a special role in cytotoxic granules: Cathepsin C processes the progranzymes to the active enzyme, and membrane-associated cathepsin B helps protect the killer cell from membrane damage in the immune synapse by perforin. Other cathepsins may substitute for these cathepsins when they are absent or mutated. |
GRANULE-MEDIATED CELL DEATH
Killer Cell Granules
Killer cells contain cytotoxic granules that are acidic, electron-dense, specialized secretory lysosomes89 (Fig. 37.2). These granules are mobilized like secretory vesicles in other secretory cells, such as neurotransmitter-containing vesicles near the synapses of neurons and melanin-containing vesicles of melanocytes. Cytotoxic granules contain the granzymes, trypsin-like serine proteases, whose major job is to initiate programmed cell death in cells marked for immune elimination. Cytotoxic granule proteins also regulate the survival of activated lymphocytes and may also cause inflammation by acting on extracellular substrates. The granzymes are trypsin-like serine proteases that use a classic histidine, serine, aspartic acid catalytic triad to cleave their substrates. Human granzymes A, B, C, and M; rat granzyme B; and human progranzyme K have all now been crystallized with high resolution.90,91,92,93,94,95 The active granzymes are produced by cleavage of a dipeptide from the N-terminus of the proenzyme. Activation is accompanied by a radical conformational change. Progranzyme K has a more rigid structure lacking an open active site than the active granzymes.
Detailed information about the conformation surrounding the active sites of granzyme A and B has provided the structural basis for understanding how subtle differences in the active site conformation lead to substantial differences in substrate specificity. As a consequence, mouse granzyme B is preferentially able to cleave mouse procaspase-3, whereas human granzyme B is better able to cleave the human orthologue. Granzyme A differs from the other granzymes in forming a covalent homodimer; the other granzymes are monomeric. Dimerization creates an extended site for substrate binding that is believed to confer a high degree of specificity to granzyme A for its substrates.90,96 In particular, because of the extended exosite for substrate binding, granzyme A substrates do not share a common short peptide sequence around the cleavage site.
Detailed information about the conformation surrounding the active sites of granzyme A and B has provided the structural basis for understanding how subtle differences in the active site conformation lead to substantial differences in substrate specificity. As a consequence, mouse granzyme B is preferentially able to cleave mouse procaspase-3, whereas human granzyme B is better able to cleave the human orthologue. Granzyme A differs from the other granzymes in forming a covalent homodimer; the other granzymes are monomeric. Dimerization creates an extended site for substrate binding that is believed to confer a high degree of specificity to granzyme A for its substrates.90,96 In particular, because of the extended exosite for substrate binding, granzyme A substrates do not share a common short peptide sequence around the cleavage site.
The cytotoxic granules also contain perforin, a poreforming molecule that delivers the granzymes into the target cell. Another pore-forming molecule, granulysin, which is homologous to the saposins, is cationic and selectively active at disrupting negatively charged bacterial and possibly fungal and parasite cell membranes. Granulysin is expressed in humans and nonhuman primates, and orthologues are found in some other species (pigs, cows, and horses), but not in mice. The positively charged cytotoxic effector molecules are bound in the granule to an acidic proteoglycan, called serglycin, after its many Ser-Gly repeats.97,98 In addition to these specialized molecules, the cytotoxic granules also contain lysosomal enzymes, the cathepsins, and internal lysosomal membrane proteins, such as CD107 (Lamp1). The outside of the granule membrane binds soluble N-ethylmaleimidesensitive factor accessory protein receptor (SNARE) proteins, synaptotagmins and Rab GTPases, which regulate vesicular trafficking and cytotoxic granule release. Some of these molecules, including Rab27a and Munc13-4, which are important for granule exocytosis, are only incorporated into cytotoxic granules as they mature by fusion of cytotoxic granules with specialized exocytic vesicles, formed in secretory cells by fusion of late endosomes and recycling endosomes. Some of the granule-associated molecules associate with lysosomes in all cells, whereas some have a specialized function in killer cells.
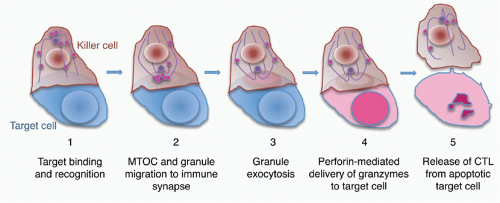 FIG. 37.3. Steps in Granule-Mediated Cytotoxicity. After the killer cell recognizes a target cell (1), an immune synapse is formed at the interface and the microtubule organizing center (MTOC) moves to the synapse, reorganizing the microtubule network (2). Cytotoxic granules move along microtubules to dock at the killer cell membrane in the central supramolecular activation complex (c-SMAC) of the immune synapse. Granule membranes fuse with the killer cell plasma membrane, releasing their contents (magenta) into the immune synapse (3). Perforin delivers the granzymes into the cytosol of target cells (4) where they initiate apoptotic death (5). The granzymes concentrate in the nucleus of target cells. The killer cell then detaches from the dying cell and is free to seek out additional targets. |
Steps in Granule Exocytosis
When CTLs and NK cells form an immune synapse with a target cell, engagement of activating receptors, including the TcR, NK cell-activating receptors, and Fc receptors, stimulates the killer cell to destroy the target cell7 (Figs. 37.3 and 37.4). Activation for cytolysis is enhanced by binding of CD8 or CD4, costimulatory receptors, and adhesion molecules such as LFA-1, which cluster in well-defined concentric rings within the immune synapse. Killer cell activation causes a calcium flux that induces lytic granules to cluster around the microtubule organizing center and then align along the immunologic synapse.99,100,101,102,103 Granules move to the immune synapse via both the microtubule network and actin cytoskeleton. The latter interaction is via myosin IIA in NK cells.104 The actin meshwork thins around the site of the synapse to make room for granules to move through it.105,106,107 Cytotoxic granules then dock to the killer cell plasma membrane in the central region of the immune synapse (c-SMAC). In T cells, granule docking and fusion may localize to a distinct (secretory) region of the central cluster (c-SMAC) of the immune synapse that is separate from the signaling domain containing the T-cell receptor and associated kinases.108 Recent studies did not observe a separation of signaling and secretory domains in the c-SMAC of NK cells. Cytotoxic granule docking is orchestrated by binding
of Rab27a on the cytosolic side of the mature granule membrane with synaptotagmin-like proteins, SLP1 or SLP2, which are anchored in the cell membrane. Docked granules are then primed for fusion by the interaction of Munc13-4 on their surface with syntaxin 11 on the killer cell membrane. This triggers the formation of a SNARE complex, the molecular machine for granule membrane fusion, between a cytotoxic granule vesicle-associated SNARE complex component (VAMP) protein with syntaxin 11 and SNAP23 on the cell membrane. Of the seven human VAMP proteins, studies in cytotoxic T cells have suggested that VAMP8 is required, whereas in NK cells both VAMP4 and VAMP7 are needed for different steps leading to granule exocytosis.109,110 Granule membrane fusion also requires participation of Munc18-2 to trigger the conformational activation of the SNARE complex. Although the general mechanism of granule exocytosis described previously is used by all killer cells, some of the details of granule trafficking and fusion at the synapse may differ between killer T cells and NK cells (although apparent differences may disappear when the same high-resolution techniques are applied to both types of killer cells). Cytotoxicity and granule fusion may occur even in the absence of a stable synapse.111
of Rab27a on the cytosolic side of the mature granule membrane with synaptotagmin-like proteins, SLP1 or SLP2, which are anchored in the cell membrane. Docked granules are then primed for fusion by the interaction of Munc13-4 on their surface with syntaxin 11 on the killer cell membrane. This triggers the formation of a SNARE complex, the molecular machine for granule membrane fusion, between a cytotoxic granule vesicle-associated SNARE complex component (VAMP) protein with syntaxin 11 and SNAP23 on the cell membrane. Of the seven human VAMP proteins, studies in cytotoxic T cells have suggested that VAMP8 is required, whereas in NK cells both VAMP4 and VAMP7 are needed for different steps leading to granule exocytosis.109,110 Granule membrane fusion also requires participation of Munc18-2 to trigger the conformational activation of the SNARE complex. Although the general mechanism of granule exocytosis described previously is used by all killer cells, some of the details of granule trafficking and fusion at the synapse may differ between killer T cells and NK cells (although apparent differences may disappear when the same high-resolution techniques are applied to both types of killer cells). Cytotoxicity and granule fusion may occur even in the absence of a stable synapse.111
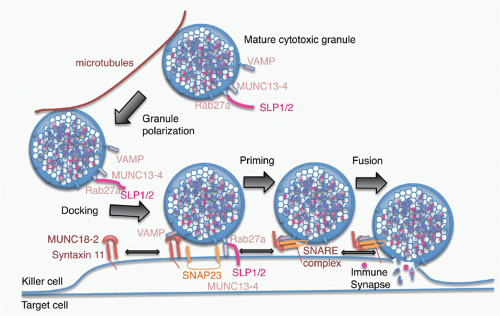 FIG. 37.4. Model of Granule Exocytosis. In response to antigen recognition, the mature cytotoxic granule moves along microtubules, probably with assistance from the actin-myosin cytoskeleton (not shown) to dock at the cell membrane at the immune synapse. A cytotoxic granule vesicle-associated soluble N-ethylmaleimide-sensitive factor accessory (SNARE) complex component (VAMP) protein binds to Munc18-2, which is associated with plasma membrane syntaxin 11. Cytotoxic granule proteins Rab27a and Munc13-4, in association with a synaptotagmin SLP1 or SLP2, help anchor the granule to the membrane. A SNARE complex forms between plasma membrane SNAP23 and syntaxin 11 and granule membrane VAMP to initiate fusion of the granule membrane to the plasma membrane. Following membrane fusion, the cytotoxic granule contents are released into the immune synapse. After fusion, granule membrane-associated cathepsin B (not shown) is displayed on the killer cell membrane and protects it from perforin membrane damage. Figure adapted from de Saint Basile et al.7 |
Genetic Diseases Caused by Defects in Perforin or Granule Exocytosis
Inherited deficiencies in perforin or the genes encoding syntaxin 11, Munc13-4, and Munc18-2 that orchestrate cytotoxic granule trafficking and release are linked to defective cytotoxicity and profound immunodeficiency.112,113,114,115,116,117,118 Patients with mutations in these genes develop the familial hemophagocytic lymphohistocytosis (FHL) syndrome. These patients are handicapped in controling viral infections and develop a severe immune activation syndrome that is often fatal in childhood unless treated with bone marrow transplantation. Some patients with milder perforin mutations that do not completely eliminate cytotoxic function are not diagnosed until adulthood. These adult patients with FHL not only have impaired antiviral immunity but are also more prone to develop lymphoma (such as perforin-deficient mice). The most prominent and sometimes fatal clinical manifestation of FHL is an inflammatory syndrome caused by uncontrolled activation and expansion of CD8 T cells, often in response to poorly controlled herpes virus infections, which leads to systemic activation of macrophages, which infiltrate tissues and overproduce proinflammatory cytokines. Macrophage activation is driven by excessive production of IFNγ by activated CD8 T cells.10,119 Sequencing
of perforin mutations in patients with FHL has identified nonsense, frameshift, and missense mutations that disrupt perforin synthesis, folding, or activity.5,120 The importance of some of these have been validated by testing cytolytic function of rat basophilic leukemia cells engineered to express mutant perforin and granzyme B. Defects in genes encoding the AP-3 adaptor—needed to shuttle cargo from the Golgi to secretory lysosomes—the lysosomal trafficking regulator LYST, or Rab27 lead to human syndromes (Hermansky-Pudlak syndrome type 2, Chediak-Higashi syndrome, and Griscelli syndrome type 2, respectively) and corresponding mouse models (pearl, beige, and ashen mice, respectively) in which cytotoxicity as well as other processes involving secretory lysosomes are defective. In fact, mice and humans with defects in these genes have defects in pigmentation due to defective melanosome transport.
of perforin mutations in patients with FHL has identified nonsense, frameshift, and missense mutations that disrupt perforin synthesis, folding, or activity.5,120 The importance of some of these have been validated by testing cytolytic function of rat basophilic leukemia cells engineered to express mutant perforin and granzyme B. Defects in genes encoding the AP-3 adaptor—needed to shuttle cargo from the Golgi to secretory lysosomes—the lysosomal trafficking regulator LYST, or Rab27 lead to human syndromes (Hermansky-Pudlak syndrome type 2, Chediak-Higashi syndrome, and Griscelli syndrome type 2, respectively) and corresponding mouse models (pearl, beige, and ashen mice, respectively) in which cytotoxicity as well as other processes involving secretory lysosomes are defective. In fact, mice and humans with defects in these genes have defects in pigmentation due to defective melanosome transport.
Lessons from Knockout Mice
Mice genetically deficient for granzymes A, B (and the granzyme B cluster), and M and perforin provide important tools for probing the importance of these effector molecules in immune defense.1 Perforin-deficient mice112 closely recapitulate the symptoms of humans with genetic perforin deficiency. They are severely immunodeficient and compromised in their ability to defend against viruses and tumors and develop the inflammatory syndrome of FHL when infected with mouse cytomegalovirus.121 Mice deficient in any 1 of the 10 granzymes, or even of the granzyme B cluster, only have subtle differences compared to wild-type animals. These experiments highlight the functional redundancy of the granzymes. While only one molecule (perforin) effectively delivers the granzymes into target cells, each of the granzymes can trigger cell death. However, target cells may be selectively resistant to one or another of the granzymes (ie, by bcl-2 overexpression or by expression of viral serpins). Requirements for a single granzyme have been shown in some cases by specific immune challenges. For example, granzyme A-deficient mice are more susceptible to the poxvirus ectromelia122 and granzyme B-deficient mice have less GvHD.123 In constructing genetically deficient mice, genetic alterations of one gene can affect the expression of nearby granzyme genes. In the original granzyme B knockout mice, the PGK-neo cassette remaining in the granzyme B locus impedes the expression of other granzyme cluster genes (granzymes C, D, and F). The granzyme B gene has also been deleted keeping the expression of granzymes C, D, and F intact.124 Cytotoxic T cells from the granzyme B-specific deletion mouse are significantly more effective at inducing apoptosis than those from the granzyme B-cluster knockout animal, underlining the importance of the other granzyme B cluster granzymes, especially when granzyme B is absent.
Because granzyme A and B are the most abundant granzymes in T cells, granzyme A/B doubly deficient mice are more immunodeficient than the single knockouts.125,126,127 CTLs from granzyme A/B-deficient mice, although somewhat impaired in cytotoxicity relative to wild-type cells, nonetheless largely retain the ability to kill target cells.128,129,130 However, the timing of key molecular events during apoptosis, such as externalization of phosphatidylserine (annexin V staining), is delayed during cell death induced by granzyme A/B-deficient CTLs versus wild-type CTLs.131 Cytotoxic T cells lacking granzyme A and B induce a modified form of cell death that seems morphologically distinct from either perforin-mediated necrosis or wild-type CTL-mediated apoptosis.131 Granzyme A/B-deficient animals do not develop spontaneous tumors and clear many viruses normally. The likely explanation of these results is that the other “orphan” granzymes (particularly H/C, K, and M),132,133,134 substitute for granzyme A and B.
Although granzyme M is highly expressed in innate immune killer cells, including NK cells, NKT cells, and γδ T cells, granzyme M-deficient mice have normal NK and T cell numbers and NK activity against tumors.135 Defense against the mouse poxvirus ectromelia and implanted NK-sensitive tumors is unimpaired in granzyme M-deficient mice compared to wild-type mice. Deficient mice are somewhat impaired in responding to mouse cytomegalovirus infection as they have higher viral levels, but they are eventually able to clear the infection. Thus, granzyme M does not appear to be essential for NK cell-mediated cytotoxicity.
Perforin Delivery of Cytotoxic Molecules into Target Cells
When the granule membrane fuses with the killer cell membrane, the granule contents are released into the synapse. Granzymes and perforin probably dissociate from serglycin in the immune synapse before they enter target cells.136 Granzymes bind to the target cell membrane by electrostatic interactions (granzymes are very positively charged and the cell surface is negatively charged)137,138,139 and also by specific receptors, such as the cation-independent mannose-6-phosphate receptor.140 However, specific receptors are not required for binding, internalization, or cytotoxicity.137,139,141,142 The lack of a receptor enables all types of cells to be eliminated and limits escape from immune surveillance. The granzymes are delivered into the target cell (but not the killer cell) by perforin where they initiate at least three distinct pathways of programmed cell death. Although perforin is essential for granule-mediated cytotoxicity to deliver the granzymes into cells, the granzymes are redundant, as each granzyme can independently activate cell death. Although genetic deficiency of one or a few granzymes does not lead to severe immunodeficiency, mice lacking one or another granzyme display subtle differences in their ability to control specific viral infections. Why are there so many granzymes? The immune system needs to contend with a wide variety of tumors and infections, some of which have elaborated strategies to evade apoptosis and immune destruction. Some of the granzymes may have evolved to disarm specific intracellular pathogens. The interplay between granzyme B and H and adenovirus illustrates how multiple granzymes may have evolved to eliminate important pathogens.143,144,145 Although both enzymes can cleave and inactivate at least two adenoviral proteins, the virus has also developed a way of inactivating granzyme B. Granzyme H potentiates the effect of granzyme B by destroying an adenoviral granzyme B inhibitor.
Perforin delivers granzymes and other effector molecules into the target cell cytosol.146,147 At high concentrations,
perforin multimerizes in a cholesterol- and calcium-dependent manner in the plasma membrane of cells to form 5- to 30-nm pores.148,149,150,151,152 Recent cross-linking and biophysical studies suggest that perforin may form at least two types of pores in membranes: small pores composed of about seven monomers that are not stable and much larger stable pores.153,154 Cryoelectron microscopy reconstructions suggest that the large pores are composed of approximately 19 to 24 subunits and have a lumen large enough for granzyme monomers or granzyme A dimers to readily pass through. The precursor of human perforin is a 555 amino acid protein synthesized with a 21 amino acid leader sequence. The N-terminal region of the mature 67-kDa protein (residues 44-410 of the human protein) is homologous to domains in complement proteins C6, C7, C8a, C8b, and C9 that form the complement membrane attack complex (MAC). The crystal structure of monomeric mouse perforin was recently solved.152 The complement homology domain, termed the MAC/perforin domain (MACPF), is similar in structure to that of bacterial pore-forming cholesteroldependent cytolysins, although they insert into membranes in opposite orientations. The MACPF domain of perforin is followed by an epidermal growth factor (EGF)-like domain; a C2 domain, a domain present in synaptotagmins and other calcium-dependent proteins, which becomes able to bind to lipid membranes after a conformational change in response to calcium; and a short 12 amino acid C-terminal peptide. The docking of the calcium-bound C2 domain is the first step in pore formation. Docking likely triggers both multimerization and a major conformational change in which two clusters of α-helices in the MACPF domain jackknife into the membrane. It is unclear whether multimerization to form a pore occurs before or after this conformational change. Perforin is glycosylated at two sites: one in the MACPF domain and one in the C-terminal peptide. Glycosylation of at least one site is needed for targeting perforin to cytotoxic granules, probably via binding of the glycan to the mannose-6-phosphate receptor.155 En route to or in the granule, the glycosylated C-terminal peptide is removed from human (but not mouse) perforin by an undefined cysteine protease to produce the mature active protein.156
perforin multimerizes in a cholesterol- and calcium-dependent manner in the plasma membrane of cells to form 5- to 30-nm pores.148,149,150,151,152 Recent cross-linking and biophysical studies suggest that perforin may form at least two types of pores in membranes: small pores composed of about seven monomers that are not stable and much larger stable pores.153,154 Cryoelectron microscopy reconstructions suggest that the large pores are composed of approximately 19 to 24 subunits and have a lumen large enough for granzyme monomers or granzyme A dimers to readily pass through. The precursor of human perforin is a 555 amino acid protein synthesized with a 21 amino acid leader sequence. The N-terminal region of the mature 67-kDa protein (residues 44-410 of the human protein) is homologous to domains in complement proteins C6, C7, C8a, C8b, and C9 that form the complement membrane attack complex (MAC). The crystal structure of monomeric mouse perforin was recently solved.152 The complement homology domain, termed the MAC/perforin domain (MACPF), is similar in structure to that of bacterial pore-forming cholesteroldependent cytolysins, although they insert into membranes in opposite orientations. The MACPF domain of perforin is followed by an epidermal growth factor (EGF)-like domain; a C2 domain, a domain present in synaptotagmins and other calcium-dependent proteins, which becomes able to bind to lipid membranes after a conformational change in response to calcium; and a short 12 amino acid C-terminal peptide. The docking of the calcium-bound C2 domain is the first step in pore formation. Docking likely triggers both multimerization and a major conformational change in which two clusters of α-helices in the MACPF domain jackknife into the membrane. It is unclear whether multimerization to form a pore occurs before or after this conformational change. Perforin is glycosylated at two sites: one in the MACPF domain and one in the C-terminal peptide. Glycosylation of at least one site is needed for targeting perforin to cytotoxic granules, probably via binding of the glycan to the mannose-6-phosphate receptor.155 En route to or in the granule, the glycosylated C-terminal peptide is removed from human (but not mouse) perforin by an undefined cysteine protease to produce the mature active protein.156
The original model for how perforin delivers granzymes into cells was that granzymes entered cells through perforin pores in the target cell plasma membrane. This model predicts that granzymes directly pass and disperse into the target cell cytosol. However, granzymes do not directly enter the cytosol but instead are first endocytosed into clathrincoated vesicles and transported to endosomes.154,157,158 Thus, the original model is not correct. Current data suggest that perforin indeed forms target cell plasma membrane pores, but these pores are small and transient (Fig. 37.5). However, calcium flows into the target cell through these pores and remains elevated for a few minutes. Because intracellular calcium is low in cells with an intact cell membrane, the cell senses a calcium influx as a sign of disruption of the plasma membrane. The elevated calcium triggers a cellular membrane damage response (also known as cellular wound healing) in which intracellular vesicles move to the plasma membrane and fuse their membranes to patch holes, and any damaged membrane is rapidly removed and internalized into endosomes.158,159,160,161 At the same time, membrane-bound granzymes, granulysin, and perforin are endocytosed. Elevated cytosolic calcium activates endosomal fusion, and granzyme- and perforin-containing endosomes fuse to form giant endosomes approximately 10 times larger than normal endosomes that have been termed gigantosomes. In the endosomal membrane, perforin forms larger and more stable pores through which granzymes begin to leak out into the cytosol. About 15 minutes after cell death has been triggered, the gigantosomes rupture, releasing any remaining cargo to the cytosol where they begin to activate programmed cell death. When the membrane repair response is inhibited, because the cell remains leaky, target cells die by necrosis instead of by slower, regulated, and energy- dependent programmed cell death.
Although perforin is the major molecule responsible for granzyme delivery, under some circumstances other molecules might serve that function. For example, bacterial and viral endosomolysins can substitute for perforin in vitro (and are widely used as laboratory reagents for intracellular delivery162) and potentially might play a similar role in vivo. The heat shock protein (Hsp)70, which chaperones some peptides across cell membranes, can also carry granzyme B (and presumably other granyzmes) into cells.163 Hsp70 is on the surface of some stressed cells or tumor cells and might help to remove these cells from the body.
Programmed Cell Death Pathways Activated by Granzymes
Once in the cytosol, the granzymes independently activate several parallel pathways of programmed cell death6 (Table 37.1). Granzyme B cleaves and activates the caspases and also directly cleaves many important caspase substrates. Granzyme B can activate cell death that mimics caspase activation, even when the caspases are inhibited or in cells in which the caspase mitochondrial pathway is deficient. Granzyme A activates a distinct programmed cell death pathway that does not involve the caspases or disrupt the mitochondrial outer membrane. The substrates of the two major granzymes are largely nonoverlapping. The exceptions, lamin B and PARP-1, may indicate common features needed for cells undergoing all forms of programmed cell death, such as disruption of the nuclear membrane, inhibition of deoxyribonucleic acid (DNA) repair, or maintaining cellular adenosine triphosphate levels. What is known about cell death executed by the other (so-called orphan) granzymes is briefly described in the following. The orphan granzymes may be more highly expressed under conditions of prolonged immune activation.164 The orphan granzymes are functionally important as mice genetically deficient in the whole granzyme B cluster are less efficient at clearing allogeneic tumors than mice deficient in just granzyme B.124 Although some key granzyme proteolytic substrates are in the cytosol (ie, Bid, caspase-3, and ICAD for granzyme B), other important targets are in other membrane-bound cellular compartments, especially the nucleus and mitochondrion. In the cytosol, granzymes B, H, and possibly K also directly cleave the proapoptotic BH3-only Bcl-2
family member Bid to initiate the classical mitochondrial apoptotic pathway that leads to mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c and other proapoptotic proteins from the intermembrane space.165,166,167,168,169,170,




family member Bid to initiate the classical mitochondrial apoptotic pathway that leads to mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c and other proapoptotic proteins from the intermembrane space.165,166,167,168,169,170,
Related posts:
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
