Carcinogenesis from the Perspective of Targeted Therapy and Immunotherapy
Introduction
In this section, we will discuss elements of cellular biology and immunology that have given insight into personalized genomically based treatment and fueled a revolution in cancer management.
Cancer represents a group of heterogeneous diseases ranging from slow to fast growing, low to high metastatic potential, and little to virulent lethality. Cancers are perverse “pseudo-organs” that, for the most part, are not a static collection of homogeneous cells, but rather a dynamic process (i.e., carcinogenesis).
Starting with the groundbreaking work of Garth Nicholson on the model of cell membranes—the Fluid Mosaic Model, architecture of normal and malignant cells—researchers have recognized a relation between the extent of chromosomal mutation, variation or instability in repetitive short DNA sequences (microsatellites, aka MSI, or short tandem repeats), and genetic and immunologic heterogeneity. Examples of MSI are TATATATATATA (dinucleotide) or GTC GTC GTC GTC GTC GTC (trinucleotide microsatellite). These motifs act as “fillers” and do not have coding function, but they are important in terms of “genetic fingerprinting,” forensic medicine, and paternity testing; involve the risk of developing cancer; and are thought to affect the likelihood of response to immunotherapy and checkpoint inhibitors (Krontiris et al., 1993; Singer and Nicolson, 1972).
Clonogenic elements, together with the inflammatory and stromal mesenchymal elements, “hijack” many of the normal processes of inflammation and tissue repair, produce an entire tumor microenvironment, parasitize the host, and then create life-threatening complications. Malignant functions are acquired in many ways and at various points in the cell cycle.
Hanahan and Weinberg, in their seminal 2011 article “Hallmarks of Cancer: The Next Generation,” provide an enlightening interpretation, on which our understanding continues to build. It is useful to consider five common themes in the initiation of carcinogenesis. These are also shown in Figure 2.1.
Individual Genetic Risk. Genetic predisposition is either due to sporadic or familial germ-line traits or due to decreased or defective DNA damage repair with loss of tumor suppressor function. These include BRCA-1 and -2-related breast and ovarian cancers, familial colon cancers or melanomas, and other cancer families (see familialcancerdatabase.nl).
Carcinogens. Carcinogens are ubiquitous in the environment and lead to mutations, chromosomal translocations, deletions, or fusion products that produce uncontrolled growth unless the
cellular surveillance and repair systems remove or fix them. An underappreciated aspect includes continued promotion by prolonged exposure. For example, among patients with tobacco-associated lung cancer, small cell cancer is frequently seen in very heavy long-term smokers. This forms another rationale for a benefit to smoking cessation even in patients with metastatic cancer (Davis et al., 2009).
Chronic Inflammation. Inflammation and the presence of lymphocytes, granulocytes, macrophages, mast cells, and their cytokines provide growth factors that appear to intensify cancer development.
Proliferative Advantage. Cellular metabolism becomes reprogrammed to support the energy needs for immortalized cell growth and give cancer cells a deadly proliferative advantage.
Evasion of Immune Surveillance. Malignant cells employ a variety of camouflage tactics to evade the immune surveillance system and allow the cancer to grow and spread.
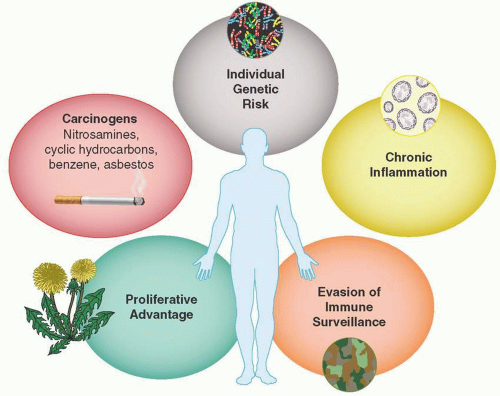 Figure 2.1 |
Processes Cancers Use to Grow
This section reviews carcinogenesis from the perspective of targeted therapy. Great progress has occurred in defining the key processes cancers use to grow and develop into life-threatening diseases, perhaps most notably by Hanahan and Weinberg (2011). The hope has been to find driver mutations that can be controlled by one or more specific, less toxic-targeted agents.
Unfortunately, cancers are nimble, have the ability to change their behavior, have many redundant pathways, and often develop resistance to even the most elegant gene-based therapy. Here, we review initial genomic and metabolic changes; uncontrolled growth, promotion, and maintenance of the primary tumor; tumor spread; and acquired resistance to targeted therapy. Focusing on these important cellular defects may reveal new paths for cancer treatment. Fourteen important processes that contribute to the growth of cancers are shown in Figure 2.2.
CELL SIGNALING AND THE DEFECTS CONTRIBUTING TO CARCINOGENESIS
It is humbling, indeed, to realize that even a simplified diagram of the “big picture” of cell signaling consists of numerous types of membrane receptors, cytoplasmic systems, and nuclear factors (see Signal Transduction—The Big Picture on page xx). These systems and the cell cycle regulators are discussed in Section 3.
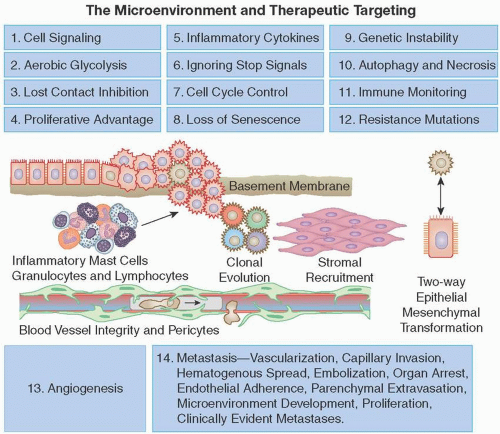 Figure 2.2 |
METABOLISM, GLYCOLYSIS, AND THE WARBURG HYPOTHESIS
Normal energy production uses glucose in the cytoplasm and pyruvic acid in the mitochondria. Under anaerobic conditions, glycolysis predominates. Otto Warburg (1930, 1956a, 1956b) first observed that cancer cells can carry out “aerobic glycolysis” to get more glucose into the cell and compensate for the much lower efficiency of ATP production than that by mitochondrial oxidative phosphorylation.
The basis of the positron emission tomography (PET) scan is that cancer cells can sequester fluorodeoxyglucose and “light up” the cancer. It is an example of how cancers achieve a proliferative advantage. This is accomplished at least in part by upregulating glucose transporters such as GLUT1 (Jones and Thompson, 2009). Glycolysis is also upregulated by RAS oncoprotein and hypoxia, which drive up levels of HIF1α and HIF2α transcription factors (Semenza, 2010a, 2010b).
Excess glycolysis also allows glycolytic intermediates to incorporate nucleosides and amino acids that are needed for the growth of new cells. Some cancers actually have a hybrid complementary energy system: glucose-dependent (“Warburg effect”) cells that secrete lactate and other cells that use the lactate and the citric acid cycle (Kennedy and Dewhirst, 2010). This flexibility is particularly helpful because tumors can have fluctuating degrees of hypoxia due to metabolic instability and abnormal neovasculature.
Gliomas and other human cancers can develop gain-of-function mutations in the isocitrate dehydrogenase 1/2 (IDH) enzymes, producing elevated oxidation and stability of the HIF1 transcription factors (Reitman and Yan, 2010; Yen et al., 2010). Glutamine (GLU) is a nonessential amino acid that is consumed by proliferating cells far more than other amino acids. Glutaminase removes an amide group that produces glutamic acid, which, in turn, loses its amine group to form alpha-ketoglutarate.
Ultimately, GLU contributes to the supply of oxaloacetate, citrate, NADPH, and carbon molecules—all critical to mitochondrial energy production. GLU is sequestered in the cell by solute carrier (SLC) transporters and serves as a nitrogen donor for nucleotide synthesis mediated by the c-MYC oncogene. GLU also activates mTORC1, the mammalian target of rapamycin complex 1. Taken together, these observations form a strong argument that cancer cells can be highly GLU dependent (“addicted”) and inhibiting GLU-mediated metabolism in the cancer cells can be a highly effective therapeutic tactic (Wise and Thompson, 2010).
LOSS OF CONTACT INHIBITION
Normal tissue growth and repair depends on cells responding to negative growth signals and knowing when to stop when they come into contact with each other. Keloids are macroscopic examples of wounds that “heal too much” when this recognition does not occur. One of the earliest microscopic signs of cell transformation is this “heaping up” of cells (Figure 2.3).
There are several ways in which normal cells respond to negative growth signals. Merlin, STK11, and TGFb are three examples of growth controls that may be mutated in various cancers (Hanahan and Weinberg, 2011). Merlin, the neurofibromatosis 2 (NF2) gene product, promotes contact inhibition and cell-to-cell bonds by coupling cell surface adhesion molecules (e.g., E-cadherin) to transmembrane receptor tyrosine kinases (RTKs) (e.g., the epidermal growth factor receptor [EGFR]) and blocks mitogenic signals (Curto et al., 2007).
Serine/threonine kinase 11 (STK11), also known as liver kinase B1 (LKB1) or renal carcinoma antigen NY-REN-19, is an epithelial polarity protein that maintains epithelial structural integrity. Germ-line mutations in this gene have been associated with Peutz-Jeghers syndrome, an autosomal dominant disorder characterized by the growth of polyps in the gastrointestinal tract, pigmented macules on the skin and mouth, and other neoplasms. Somatic mutations of the LKB1 gene have been found in lung, cervical, breast, intestinal, testicular, pancreatic, and skin cancers. LKB1 can block MYC oncogene function (Partanen et al., 2009).
Transforming growth factor beta (TGF-β) is a multipurpose cytokine activity that exists in three isoforms. It is part of a superfamily that includes inhibins, activin, anti-Müllerian hormone, bone morphogenic protein, decapentaplegic, and Vg-related protein 1. TGF-β is known for its antiproliferative effects (Ikushima and Miyazono, 2010). However, in many late-stage tumors, TGF-β signaling changes from suppression of cell proliferation and instead activates a cellular program, termed the epithelial-to-mesenchymal transition (EMT) (see EMT and Metastasis on p. xx.), seen in high-grade malignancy.
Cancer as a “Pseudo-Organ” with Great Proliferative Advantage
ORCHESTRATING THE DEVELOPMENT OF THE TUMOR MICROENVIRONMENT
Displaying a useful graphic depiction of the highly complex network of microenvironmental signaling interactions remains a great challenge, made even more difficult due to tumor progression and the
interplay between neoplastic and supporting stromal cells. This model becomes even more complex as cancers metastasize.
interplay between neoplastic and supporting stromal cells. This model becomes even more complex as cancers metastasize.
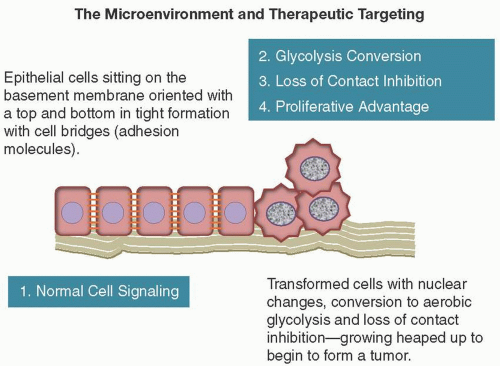 Figure 2.3 |
Cancer Stem Cells
Cancer stem cells (CSCs) are the “seeds” that develop when normal tissue stem cells undergo transformation. CSCs are often more resistant to chemotherapy and radiation and may also become dormant only to recur even years after initial treatment. What starts as a “clone then may become genetically and phenotypically heterogeneous with variable degrees of differentiation” (Nguyen et al., 2012).
Genome sequencing has revealed striking intratumoral genetic diversity within adjacent areas in the same tumor deposit (Gerlinger et al., 2012). This plasticity helps explain the frequent mutation to resistance seen following dramatic responses to targeted therapy in EGFR-mutated lung cancer and the need for different strategies for what appears to be the identical V600E BRAF mutation that is often sensitive to a single agent in melanoma but not as often beneficial in colorectal cancer.
Even in the area of BRAF-mutated metastatic melanoma, one of the great oncology success stories, not all patients have a durable benefit. One variable in the therapeutic equation is NRAS, which can affect the response to drugs such as vemurafenib (Kelleher and McArthur, 2012).
Endothelial Cells
Endothelial cells form a thin layer on the interior surface of blood and lymphatic vessels throughout the circulatory system. They play a critical role in vascular biology, controlling leakage of proteins and blood cells, protecting against clots, controlling vasoconstriction and dilatation, and supporting the repair and renewal of damaged vessels (Goligorsky and Salven, 2013).
Endothelial cells contain networks of interconnected signaling pathways involving ligands of signal-transducing receptors such as the following:
Notch, a ubiquitous signaling system found in most multicellular organisms. Notch controls key branch points in embryogenesis and was first discovered as an indentation in the wings of fruit flies in 1914 by John S. Dexter (Morgan, 1917) and sequenced in the 1980s (Kidd et al., 1986).
Neuropilin is a membrane-bound coreceptor for Vascular endothelial growth factor (VEGF) and semaphorins (semaphorins are the 500-amino-acid proteins that bind to plexins and direct axon growth and neurologic development). Neuropilin is encoded by the NRP-1 gene.
Roundabout homolog 1, a membrane protein controlling axon guidance and cell adhesions, encoded by ROBO1, a member of the immunoglobulin gene superfamily
Ephrin type A receptor (encoded by the EPHA1 gene), a mediator of nervous system development implicated in carcinogenesis
VEGF. In 1983, Dr. Harold Dvorak and colleagues first showed that tumor cells secreted vascular permeability factor (VPF) and that a blocking antibody to VPF, later named VEGF, could prevent the edema and fluid accumulation that are characteristics of human cancers. VEGF is in part responsible for the abnormal vasculature seen in human tumors (Senger et al., 1983).
Angiopoietins 1/2, the ligands for the tyrosine kinase immunoglobulin/EGF receptors TIE1 and TIE2. They work intimately with VEGF and are proangiogenic (ANG-1) or cause vessel regression (ANG-2).
Fibroblast growth factor (FGF) signaling. FGFs, discovered in 1973, are “promiscuous” heparin-binding mitogens with a wide range of functions, including mesoderm induction, angiogenesis, keratinocyte organization, and wound healing.
Pericytes
Pericytes are specialized, mesenchymal, smooth muscle-like cells with finger-like projections wrapped around blood vessel endothelial cells (see Figure 2.2). Pericytes secrete VEGF as well as antiproliferative ANG-1 ligand for endothelial surface TIE2 receptors. Along with the vascular basement membrane, they prevent hydrostatic pressure-induced leakage and retard cancer cell entry to circulation (Gaengel et al., 2009; Raza et al., 2010).
PDGF inhibition reduces pericyte coverage of tumor vessels and destabilizes vascular integrity and function (Pietras and Ostman, 2010). Stimulating pericyte function, therefore, represents another tactic for metastasis inhibition.
Cancer-Associated Fibroblasts
Fibroblasts often make up a major portion of tumor stroma and appear like pawns on the cancer “chessboard.” Structural fibroblasts form the supporting structure of normal epithelium. Myofibroblasts express smooth muscle actin (SMA) in wounds and chronic inflammation. Both are seen in very dense fibrous (desmoplastic) cancers (Hanahan and Weinberg, 2011).
Stromal Progenitors
Mesenchymal stem and progenitor cells, labeled and tracked using green fluorescent protein (GFP), have been found to migrate from the marrow into tumors, like new recruits to the battlefield, where they may differentiate and transform further. Tumor-associated stromal cells may then be supplied by local cell growth, conscription of adjacent normal cells, or bone marrow stem cell migration. A novel combination targeted therapy such as liposomal doxorubicin, bevacizumab, and temsirolimus may be effective by attacking the nucleus (anthracycline), stromal blood vessels (VEGF inhibitor), and cell signaling (mTOR inhibitor) (Moroney et al., 2012). The cancer cell has a large repertoire of tactics to gain a proliferative advantage.
Stem cells can thrive in the absence of growth factors required for normal cells, but if exposed to stimulatory molecules either from themselves (autocrine) or from their neighbors (paracrine), growth accelerates. CSCs can sequester and use glucose far more efficiently and are bolstered by other cellular “recruits” that provide a nurturing protective fibrous infrastructure for cancer colonization. It is this “trifecta” that must be taken into account when devising new targeted therapies for epithelial malignancies because the multiple mutations seen in carcinomas such as non-small cell lung cancers (NSCLCs) with their redundant and nimble cellular systems are not likely to be amenable to long-term control with a single agent. This is in contrast, however, to other diseases such as chronic granulocytic leukemia or gastrointestinal stromal tumors (GIST), where imatinib may be effective for many years (Smith, 2011).
Inflammation and Inflammatory Cytokines
INFLAMMATION AND TUMOR PROMOTION
Virtually, every tumor deposit contains inflammatory cells ranging from sparse infiltrations detectable only with cell type-specific antibodies to dense cellular infiltrates apparent by standard light microscopy (DeNardo et al., 2010). These inflammatory cells supply a whole array of molecules: growth and survival factors, proangiogenic factors, matrix-modifying enzymes that facilitate invasion and metastasis, and inducers of EMT and reactive mutagenic oxygen species (Grivennikov et al., 2010). It is quite possible that some of the beneficial effects of chemotherapy actually may be due to decreasing the activity of sensitive, tumor-infiltrating inflammatory cells even when the cancer cells themselves have acquired chemotherapy resistance.
Leukocytes (macrophages, mast cells, neutrophils, and T/B lymphocytes) can be found in most neoplastic lesions and promote tumor progression (i.e., tumors are wounds that never heal) (Dvorak, 1986; Schäfer and Werner, 2008). In the course of normal wound healing and fighting infections, immune inflammatory cells appear transiently and regress in contrast with the chronic inflammation of cancer, which is associated with fibrosis, scarring, aberrant angiogenesis, and neoplasia (Grivennikov and Greten, 2010) (Figure 2.4).
Immune inflammatory cells can release a variety of cytokines that have either tumor-promoting or tumor-killing effects:
Epidermal growth factor (EGF)
Angiogenic growth factor (VEGF)
FGF2 (see FGF section above)
Chemokines, the 8-to-10-kD cysteine containing signaling proteins that induce movement (Greek—kinos) or chemotaxis in neighboring responsive cells. Chemokines are classified into subfamilies (CXC, CC, CX3C, and XC). They interact with G protein-linked transmembrane receptors.
Cytokines that amplify the inflammatory state
Metalloproteinases, zinc-requiring enzymes that degrade extracellular matrix proteins
Cysteine cathepsin (Greek, hepsein “boil,” kata “down”) proteases activated at low pH, usually inside lysosomes (Bergmann and Fruton, 1936)
Heparanase (HPSE), which cleaves heparan sulfate heavily glycosylated proteins (proteoglycans, HSPG), has been associated with poor prognosis. Heparan, a close relative of heparin, is made up of sulfated disaccharides and forms the blood vessel interior lining, which must be penetrated for cancer cells to metastasize hematogenously.
All of the above cytokines are potential targets for cancer therapeutic agents. Tumor-infiltrating myeloid cells, expressing macrophage marker CD11b and neutrophil marker Gr1, suppress CTL and natural killer (NK) cell activity. Recruitment of these myeloid elements may be doubly damaging by directly promoting angiogenesis and tumor progression as well as affording a means to evade immune destruction. Another strategy, therefore, may be to inhibit these promoter cells rather than the CSCs.
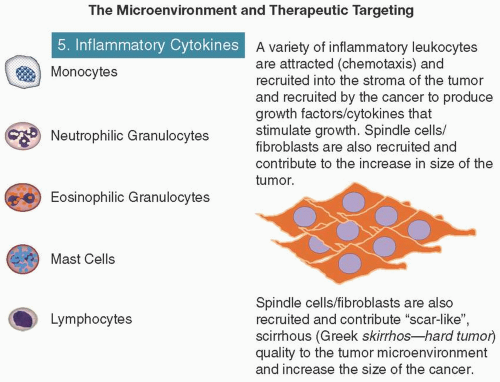 Figure 2.4 |
NECROSIS HAS PROINFLAMMATORY AND TUMOR-PROMOTING POTENTIAL
Many cancers, such as squamous cancer of the lung, demonstrate spontaneous necrosis. This is more complicated. During necrosis, in contrast to apoptosis, cells self-destruct and release their contents into the local tissue microenvironment. This results in tumor-promoting inflammation, increased angiogenesis, and proliferation due to cytokines such as IL-1α (Hanahan and Weinberg, 2011).
Cell death by necrosis appears to be under genetic control in some circumstances and, rather than being a random and undirected process, explains why some necrotic cancers can act in a very aggressive fashion and produce a whole array of paraneoplastic complications such as fever, anorexia, and fatigue (Galluzzi and Kroemer, 2008). Tumor lysis syndrome is the most extreme example of the harm that can occur when tumors die too rapidly and liberate toxic, life-threatening organic acids, potassium, and so forth. Finding ways to limit cellular necrosis could be a very effective strategy to limit cancer acceleration.
IGNORING STOP SIGNALS
Many negative feedback loops are in place to limit the growth of normal cells. These include tumor suppressors CDK-interacting protein and kinase-inhibitory protein (CIP and KIP) and inhibitor of kinase 4/alternative reading frame (INK4a/ARF). The CIP/KIP family includes the genes p21, p27, and p57. They halt cell cycle in G1 phase, by binding to, and inactivating, cyclin-CDK complexes. p21 is activated by p53 (which, in turn, is triggered by DNA damage, e.g., due to radiation). p27 is activated by TGF-β, a growth inhibitor. The INK4a/ARF family includes p16INK4a, which binds to CDK4 and arrests the cell cycle in G1 phase, and p19ARF, which prevents p53 degradation. However, these “stop signals” can be ignored by cancer cells (Figure 2.5), leading to unregulated growth (e.g., RAS mutations with decreased RAS GTPase activity in cellular events that are constantly in the “on” position rather than temporary and short lived) (Wertz and Dixit, 2010).
LOSS OF SENESCENCE AND ACQUISITION OF “IMMORTALITY”
From the early days of cell culture research, it was noted that normal cell lines are able to pass through only a limited number of successive cell growth and division cycles. This natural aging—senescence—is associated with distinct processes and entrance into a nonproliferative but viable
state. Cytoplasm enlarges, cells lose proliferation markers and accumulate lysosomal beta-galactosidase (Collado and Serrano, 2010). Some cancer cells can disable their senescence- or apoptosis-inducing circuitry.
Crisis is a term for cell death due to genetic damage (RB1, p53) or nutrient deprivation. Telomeres, composed of multiple tandem hexanucleotide repeats, shorten progressively in cultures of nonimmortalized cells. Eventually, the DNA fuses into nonviable dicentric chromosomes. The lost protective function triggers entrance into crisis.
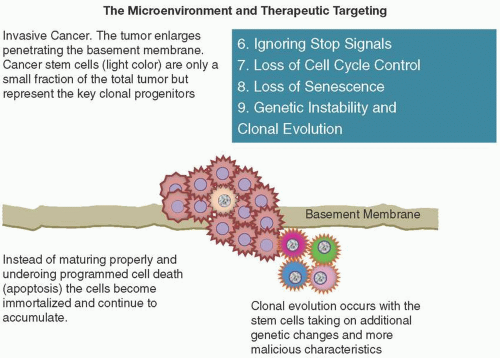 Figure 2.5 |
Telomerase, the specialized DNA polymerase, which adds repeat segments to the ends of telomeric DNA, is low or absent in normal cells but is expressed at significant levels in human cancer cells.
SENESCENCE AS A TARGET FOR CANCER THERAPY
Senescence is one way that cancer cells may “hibernate.” Senescence limits tumor progression and could represent a strategy to increase survival using tactics such as TP53 reactivation, inhibition of c-MYC in addicted tumors, or treatment with cyclin-dependent kinase (CDK) inhibitors.
INTERPLAY BETWEEN TP53 AND TELOMERASE
The loss of TP53-mediated surveillance of genomic integrity may permit cells to survive chromosomal breakage-fusion-bridge (BFB) cycles, which increase genome mutability. Delayed acquisition of telomerase function first serves to generate tumor-promoting mutations, whereas its later activation stabilizes the mutant genome and then confers “immortality.” This is another example of the “bipolar” nature of cellular processes whereby early on a cancer may develop as a result of the absence of a certain function and later the cancer may be promoted by the acquisition of that same function.
ADDITIONAL FUNCTIONS OF TELOMERASE
Telomerase and its catalytic reverse transcriptase (hTERT) are found at multiple sites along chromosomes, not just at the telomeres. This results in other functions: telomerase is a cofactor of beta-catenin/LEF transcription and amplifies WNT signaling (Park et al., 2009). It also can enhance cell proliferation, RNA polymerase, resistance to apoptosis, and DNA damage repair
(Maida et al., 2009). This recurring theme of multifunctionality adds complexity to the design of new clinical strategies but could also be exploited to great effect because a telomerase inhibitor could attack cancers in a variety of ways. A number of agents have been developed to inhibit telomerase activity and/or modulate its expression: imetelstat (GRN163L 13-mer oligonucleotide), tertomotide (GV1001 peptide vaccine), BIBR15329 (nucleoside inhibitor shortening TTAGGG repeats) (Puri and Girard, 2013). To date, however, early trials have not identified a setting in which this therapeutic strategy has produced tangible consistent benefit.
(Maida et al., 2009). This recurring theme of multifunctionality adds complexity to the design of new clinical strategies but could also be exploited to great effect because a telomerase inhibitor could attack cancers in a variety of ways. A number of agents have been developed to inhibit telomerase activity and/or modulate its expression: imetelstat (GRN163L 13-mer oligonucleotide), tertomotide (GV1001 peptide vaccine), BIBR15329 (nucleoside inhibitor shortening TTAGGG repeats) (Puri and Girard, 2013). To date, however, early trials have not identified a setting in which this therapeutic strategy has produced tangible consistent benefit.
GENETIC INSTABILITY
Multistep tumor progression can be portrayed as a sequence of clonal evolution. Given the billions of mitotic events that occur in our body, it is no surprise that additional enabling mutations can occur by chance in an already vulnerable DNA “portfolio.” In addition to frank mutation, tumor suppressor genes can be inactivated through “epigenetic” DNA methylation switching and histone modifications (Berdasco and Esteller, 2010). The extraordinary ability of genome maintenance systems to detect and repair defects in the DNA ensures that rates of spontaneous mutation are usually very low during each cell generation (Hanahan and Weinberg, 2011); however, in the course of acquiring the multiple mutant genes needed to orchestrate tumorigenesis, cancer cells often increase the rates of mutation (Negrini et al., 2010; Salk et al., 2010). These genetic changes are brought about through a breakdown in the genomic maintenance machinery.
In addition, the accumulation of mutations can be accelerated by compromising the surveillance systems that normally monitor genomic integrity and force genetically damaged cells into either senescence or apoptosis (Jackson and Bartek, 2009) (Figure 2.5). The role of TP53 is central here, leading to its being called the “guardian of the genome” (Lane, 1992). Tumor suppressors detect and repair damaged DNA. They also inactivate or intercept mutagenic molecules before they have damaged the DNA (Ciccia and Elledge, 2010; Negrini et al., 2010).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


