rational strategy to select promising agents for clinical trials through a stepwise approach of preclinical in vitro testing followed by in vivo screening.8,9,10 This system involves several phases: biochemical prescreening assays, in vitro efficacy models, in vivo short-term screening, animal efficacy testing, and preclinical toxicology testing.
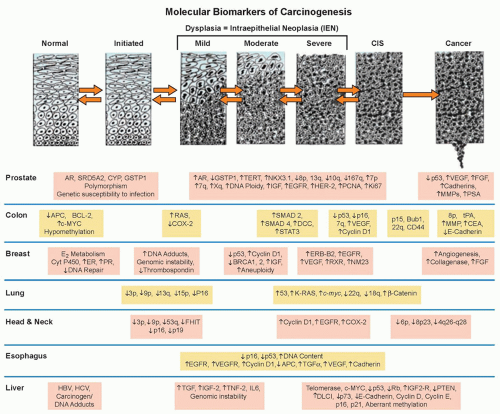 Figure 33.1 Genetic progression in major cancers. Carcinogenesis is driven by genetic progression. This progression is marked by the appearance of molecular biomarkers in distinctive patterns representing accumulating changes in gene expression and correlating with changes in histologic phenotype as cells move from normal through the early stages of clonal expansion to dysplasia and finally to early invasive, locally advanced, and metastatic cancer. The figure257 shows candidate molecular biomarkers of genetic progression in seven target organs: the prostate,300,301,302 the colon,2 the breast,303,304 the lung,305,306,307 the head and neck,308,309,310,311 the esophagus,312,313 and the liver.314 CIS, carcinoma in situ; AR, androgen receptor; CYP, cytochrome P-450; GSTP1, glutathione S transferase P1; TERT, telomerase reverse transcriptase; NKX3.1, NK 3 transcription factor related, locus 1 (prostate specific, androgren regulated); IGF, insulin-like growth factor; EGFR, epidermal growth factor receptor; HER-2, human epidermal growth factor receptor-2; PCNA, proliferating cell nuclear antigen; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; MMP, matrix metalloproteinase; PSA, prostatespecific antigen; APC, adenomatous polyposis coli; BCL-2, B-cell lymphoma 2 gene, apoptosis control; c-MYC, v-myc avian myelocytomatosis viral oncogene homolog; COX-2, cyclooxygenase-2; SMAD, homolog of mothers against decapentaplegic + C. Elegans SMA protein; DCC, deleted in colon cancer gene; CEA, carcinoembryonic antigen; ER, estrogen receptor; PR, progesterone receptor; ERB-B2, Receptor tyrosine-protein kinase erbB-2, same as HER-2; RXR, retinoid X receptor; NM23, Nucleoside disphosphate kinase A; K-RAS, Kirsten rat sarcoma viral oncogene homolog; FHIT, fragile histidine triad protein; TGFα, tumor growth factor alpha; HBV, hepatitis B virus; HCV, hepatitis C virus; TNF-2, tumor necrosis factor 2; IL6, interleukin 6; PTEN, phosphatase and tensin homolog. (Figure and revised caption from Kelloff GJ, Lippman SM, Dannenberg AJ, et al. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer—a plan to move forward. Clin Cancer Res 2006;12:3661-3697, published with permission from the American Association for Cancer Research.) |
The assays measure the ability of potential cancer risk-reducing agents to reverse transformation in normal epithelial cells exposed to carcinogens. For example, after treatment with a carcinogen such as 7,12-demethylbenz(a)anthracene, MMOCs develop lesions similar to alveolar nodules that are considered precancerous in mouse mammary glands in vivo.11 Pretreatment of organ cultures before carcinogen exposure measures the effect of cancer risk-reducing agents in the initiation stage of carcinogenesis, whereas treatment after carcinogen exposure measures activity during tumor promotion. Three of these assays (using rat tracheal epithelial, A427, and MMOC cells) have shown predictive values of 76% to 83% for cancer risk-reducing agent efficacy in in vivo models.8
TABLE 33.1 Molecular Mechanisms Common to Transforming Cells and Potential Preventive Interventions | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
TABLE 33.2A Chemical Carcinogenesis Models Used for Screening of Cancer Risk-Reducing Agents for Common Epithelial Neoplasms in Animals | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
cancer risk-reducing agents create the following challenges to be overcome: (1) the need for large therapeutic index (doses associated with potential toxicity of an intervention need to substantially exceed doses aimed at delaying or reversing transformation) for use in individuals who are asymptomatic yet may benefit from an extended (years) treatment course; (2) the long latency to malignant transformation (an assessment of effectiveness based on the reduction in cancer incidence requires studies lasting for years and involving thousands of participants); (3) adherence (once-daily dosing regimens using interventions that have sufficiently long half-lives may minimize the impact of a missed dose yet maintain the biologic impact on the physiologic target; minimal toxicity and strong psychological commitment to preventive goals also enhance adherence15); and (4) complex risk assessment for cancer (individuals with highly penetrant but infrequent, germ-line genetic susceptibility to breast and colon cancers16,17 are excellent candidates for cancer risk-reducing agents and are likely to accept some toxicity for reduced cancer risk). For individuals at more modestly increased risk (e.g., long-term, current smokers; persons with a family history of cancer; women with mammographically dense breasts), quantitative risk assessment algorithms may be useful in the future to identify optimal cancer risk-reducing agents. The refinement of cancer risk calculators for breast,18 colon,19 and prostate cancer20 promises to appropriately select high-risk individuals for cancer risk-reducing agents such that anticipated benefits exceed potential risks.
TABLE 33.2B Selected Transgenic Animal Models for Carcinogenesis Evaluation | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
(e.g., proliferation, apoptotic index), biochemical, or molecular (e.g., p53, cyclin D) end points may be used as secondary end points. Preoperative or window of opportunity trials enroll subjects for brief study periods prior to obtaining tissue by a planned resection of an invasive neoplasm. Such designs permit the exploration of biomarker modulation in the invasive neoplasm and in contiguous epithelial fields proximal and distal to the invasive neoplasm.27
TABLE 33.3 Common Intraepithelial Neoplasias | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 33.4 Characteristics of Biomarkers for Use as End Points in Cancer Risk-Reducing Agent Efficacy Assessment | |
|---|---|
|
of the head and neck or lung. None of the interventions reduced second airway primary invasive neoplasms.57 The Lung Intergroup Trial randomized patients with surgically resected lung cancer to 13cRA versus placebo and found no significant differences between the two arms in second primary tumors.58 Notably, smoking status modified the effect of the 13cRA intervention, which was harmful in current smokers yet beneficial in former smokers.
TABLE 33.5 Larger, Randomized Trials of Retinoids in Human Cancer Risk Reduction with Cancer Outcomesa,b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
xeroderma pigmentosum; however, severe, acute mucocutaneous toxicity with the 13cRA occurred.63 Also, the preventive effect of the retinoid was lost after stopping retinoid therapy. In renal transplant patients, acitretin (30 mg per day) reduced the numbers of premalignant lesions, the number of patients with skin cancer, and the cumulative number of skin cancers.64
TABLE 33.6 Randomized Trials of Antioxidant Nutrients in Human Cancer Risk Reduction with Cancer Outcomesa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
patients with nonmuscle-invasive bladder transitional cell carcinoma after transurethral resection with or without adjuvant intravesical bacillus Calmette-Guérin; recurrence rates were similar in both groups.72 Another trial randomized 65 patients with biopsy-confirmed transitional cell carcinoma of the bladder to a multivitamin (recommended dietary allowance [RDA] levels) alone or supplemented with 40,000 IU retinol, 100 mg pyridoxine, 2,000 mg ascorbic acid, 400 U of α-tocopherol, and 90 mg zinc.73 The 5-year estimate of tumor recurrence was 91% in the RDA arm versus 41% in the higher-dose nutrient arm (p = 0.0014).
cancer.96 Long-term use of multivitamin supplements, which are a major source of folate and other B vitamins, has been associated with a reduction in the risk of colon cancer in some studies, including recent (postfortification) findings.97,98,99 Supporting an anticancer role of folate is that genotypes for methylene tetrahydrofolate reductase, an enzyme known to be involved in folate metabolism, predict the risk of colon cancer dependent on folate intake or status.100 Vitamin B6 has been less studied in relation to cancer than folate, but some epidemiologic studies suggest that vitamin B6 may be important for colorectal cancer.101,102 A higher risk of cancer related to deficiencies of these vitamins has been suggested for alcohol drinkers.103
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


