Cancer of the kidney is neither common enough to cause a large percentage of cancer-related deaths nor uncommon enough to be considered an “orphan” malignancy. In that context, the progress made in uncovering the genetic basis of renal cell carcinoma (RCC), its molecular pathways, and the approval of novel therapies to perturb those pathways over the last decade is indeed remarkable. Over a relatively short time, the management options for localized kidney cancer have evolved from near universal acceptance of open radical nephrectomy (RN) to the routine use of minimally invasive partial nephrectomy (PN), thermal ablation (TA), and active surveillance (AS). Concurrently, metastatic RCC has progressed from marginally treatable, with a low incidence of spontaneous and/or immune-induced durable regression, to overall response rates (complete response + partial response + stable disease) of >50% and a near doubling of cancer-specific survival. Looking forward, it is anticipated that kidney cancer will soon become a chronic disorder as we better understand the biologic heterogeneity and systemic therapies for RCC. Herein, we review the current and rapid evolution in our understanding and management of cancer of the kidney.
EPIDEMIOLOGY, DEMOGRAPHICS, AND RISK FACTORS
Kidney cancer accounts for approximately 2% of malignancies worldwide with about 300,000 cases diagnosed per year and 100,000 deaths.1 Data suggest the incidence of RCC is more common in industrialized countries, which may be related to increased incidental detection. In the United States, tumors of the kidney account for 3% to 4% of all cancer diagnoses with an estimated 65,000 cases and 14,000 deaths.2 There is a male-tofemale predominance with the lifetime risk of a RCC diagnosis of 1:69 in men and 1:116 in women.3 While kidney cancer remains predominantly a tumor of the elderly (median age at diagnosis of 65 years), the number of new kidney cancer cases appears to be rising in younger individuals.2 This may be explained by either the increasing use of noninvasive imaging in younger patients4,5,6 or perhaps true biologic differences in the disease.7 Similarly, racial differences have also been described with increased rates and decreased survival noted in African Americans and improved survival in Asian populations.8 Whether this represents differences in health-care access or disease biology is unclear.
The most commonly cited risk factors for the development of RCC include smoking, obesity, and hypertension.9 The data regarding smoking as a risk factor for RCC appear strong. In a meta-analysis evaluating 19 case control studies and 5 cohort studies, Hunt et al.10 found a 38% increased risk in current and former smokers, noting not only a dose-risk relationship, but also an abatement of risk with smoking cessation >10 years. The relationship between obesity and RCC is less well studied, although the epidemiology seems to point to a causal association. In a quantitative review of the literature between 1966 and 1998, Renehan et al.11 calculated a relative risk of 1.07 per increase in unit body mass index and concluded that nearly a third of RCC cases may be attributable to obesity. Data suggesting a stronger association of obesity and RCC in women lead to hypotheses that the relationship may be due to dysregulation of sex hormones, insulin metabolism, or the immune system.3 The relationship between hypertension and RCC is based largely upon retrospective and/or population-based epidemiologic data. In an analysis of 13 case controlled studies, Grossman et al.12 noted hypertensive patients exhibited a pooled odds ratio of 1.75 of having RCC. While there may be a relationship between severity and duration of hypertension, given the limits of the data, if such an association exists it is difficult to ascertain.3 Finally, exposure to chronic diuretics,13 nonsteroidal analgesics,14 and tricholorethylene, a cleaning agent, have all been associated with an increased risk of RCC.3
While screening for kidney and other potentially lethal diseases is enticing, a risk benefit analysis argues against it given the low overall prevalence of RCC in the general population. One large study, in which over 200,000 adults were screened for abdominal malignancy with ultrasound, found only 192 cases of RCC (0.09%).15 Screening has, therefore, been proposed in target populations, including individuals with familial RCC syndromes such as von Hippel-Lindau disease (VHL) and those on hemodialysis, who are known to be more likely to be diagnosed with RCC. In patients on dialysis, while there appears to be an increased incidence of RCC in the native kidneys or even in a transplanted allograft, this may be due to their medical follow-up. Indeed, most patients with end-stage renal disease will have been “screened” during the evaluation and management of their renal failure/allograph, making the reasons for this association difficult to dissect. This is distinctively different than an emerging population of patients with increased genetic risk for cancer, but whose kidneys have not yet been imaged. In families who carry known mutations in the genes responsible for VHL, hereditary papillary RCC, hereditary leiomyoma RCC, Birt-Hogg-Dubé syndrome (BHD), potentially tuberous sclerosis, and/or autosomal dominant polycystic kidney disease, a renal ultrasound may be an inexpensive, low-risk, and judicious means of targeted screening. The initial timing, frequency, and effectiveness of screening in these at-risk populations are not yet established.
PATHOLOGY OF RENAL CELL CARCINOMA
Pathologic classifications assist in diagnosis and prognosis, and inform therapy. Most pathologic classifications emphasize a tumor’s morphology and histology—although, increasingly they are incorporating genetic characteristics.16 There are 10 tumor subtypes in the current World Health Organization classification system for RCC (Table 37.1). The major histologic variants include clear cell, papillary, chromophobe, and collecting duct tumors, which account for 90% to 95% of renal carcinomas,17 although less common subtypes and an “unclassified” category also exist. Uncommon subtypes of RCC, not included in the World Health Organization classification, include tubulocystic carcinoma, clear cell tubulopapillary RCC, thyroid-like follicular carcinoma, and acquired cystic disease-associated RCC. The relatively rapid movement in RCC toward molecular classification follows advances in our molecular understanding of these variants and may soon supplant simple morphologic classification.
TABLE 37.1 2004 World Health Organization Classification of Sporadic Renal Cell Carcinoma with Genetic and Clinical Correlates
Type
Genetics
Clinical
ccRCC (70% to 80%)
Deletion, mutation or methylation of 3p25-26 (VHL)
Most common variant
Prognosis predicted by stage and grade
Multilocular cystic ccRCC (uncommon)
Deletion, mutation, or methylation of 3p25-26 (VHL)
Variant of ccRCC
Distant metastases uncommon
Papillary RCC (10% to 15%)
Gain of 7 or 17 (trisomy or tetrasomy), loss of Y, deletion of 9p. Mutations of 7q31 when associated with hereditary papillary RCC
10% to 15% of RCC
95%+ 5-year cancer-specific survival in type I papillary RCC
Response to tyrosine-kinase inhibitors less robust
Chromophobe RCC (3% to 5%)
Extensive chromosomal loss of Y, 1, 2, 6, 10, 13, 17, 21 Mutations of 17p11.2 when associated with BHD
5% of RCC
Affects men and women equally with overall excellent prognosis
Collecting duct carcinoma (Bellini tumor) (<1%)
Highly variable
Losses of 1q, 6p, 8p, 9p, 13q, 19q, 21q
Male preponderance (2:1)
Mean age 55
Microscopically high grade, may resemble urothelial spectrum of cancers,
Overall poor prognosis
Renal medullary carcinoma (rare)
Not defined
Associated with sickle cell trait
Aggressive and lethal within 12 mo
Mean age 19 y
Male>female
Xp11 translocation carcinoma (rare)
Translocation of TFE3 gene on XP11.2
Children and young adults
May present at advanced state and act more aggressively in adults
Renal carcinoma associated with neuroblastoma (rare)
Not defined
Morphologically and microscopically similar to ccRCC
Mucinous tubular and spindle cell carcinoma (rare)
Adapted from Deng FM, Melamed J, Zhou M. Pathology of renal cell carcinoma. In: Libertino JA, ed. In Renal Cancer: Contemporary Management. New York: Springer; 2013:51-69.
Other pathologically relevant variables in RCC include nuclear grade, sarcomatoid and rhabdoid differentiation, tumor necrosis, and vascular invasion. Nuclear grade usually follows the Fuhrman grading system18 and is most often and best used for clear cell RCC (ccRCC) as the prognostic value in non-ccRCC remains largely unproven. Sarcomatoid differentiation exists in 5% of RCC and can be seen in any subtype. As such it is not considered a distinct entity, but rather a high-grade or poorly differentiated component. The presence and extent of micro- or macronecrosis has been correlated with prognosis in ccRCC.19 Micro- or macrovascular invasion is thought to be a requisite step toward systemic disease; however, correlation between the extent of invasion and prognosis remains imprecise.
DIFFERENTIAL DIAGNOSIS AND STAGING
Most patients with RCC present with an incidental, radiographically detected renal mass (Fig. 37.1). While symptoms including microscopic or gross hematuria, flank pain, gastrointestinal disturbances and/or pain, bleeding or systemic disturbances related to metastases may lead to the diagnosis, the use of routine cross-sectional imaging has led to the more common scenario of an incidentally detected renal mass. While the suspicion for RCC may be high in cases such as these, RCC is a pathologic/tissue diagnosis, not a clinical one (Fig. 37.2). Proper radiographic evaluation of a renal mass requires a pre- and postcontrast computed tomography or magnetic resonance imaging (MRI) to assess enhancement.20 Duplex ultrasound, renal mass biopsy, and noncontrast diffusion-weighted MRI with antibody drug conjugates mapping may be useful adjunctive tests in various clinical settings. deoxy-2[18F]fluoro-d-glucose positron emission tomography exhibits a low sensitivity for the diagnosis of RCC and is therefore not recommended for the evaluation of RCC. ImmunoPET with G-250 using an iodine-labeled antibody against carbonic anhydrase IX (CA-IX), which is known to be overexpressed in ccRCC, exhibits near 90% sensitivity and specificity for this RCC subtype.21
The differential diagnosis of the renal mass is broad and includes a long list of benign, malignant, and inflammatory conditions. Clinical and radiographic features can assist the astute clinician in narrowing down the diagnosis of the renal mass, particularly for benign and inflammatory lesions. Cystic lesions, for example, are frequently benign,22 and fat-containing solid lesions are most commonly found to be angiomyolipomas (also benign). About 20% of enhancing renal masses and 15% of surgically removed masses are nonmalignant, with the most common diagnoses being oncocytoma and fat-poor angiomyolipoma.23,24,25 Young to middle-aged women, in particular, are more likely to have benign pathology, as high as 40% in some series, while the likelihood of malignancy gradually decreases with age in men.24,26 Tumor size is the most important determinant of pathology and biologic aggressiveness with larger tumors more likely to be high grade, locally invasive, and/or of adverse histologic subtype.23,24,27 Incorporation of readily available features can allow the physician to provide an individualized risk of cancer (ranging between ˜50% and ˜99%), but a certain diagnosis requires pathologic confirmation.24,28 The most accurate nomogram currently available gives estimates of preoperative prediction of tumor histology with an area under the curve of 0.76 and high-grade malignancy with an area under the curve of 0.73.28
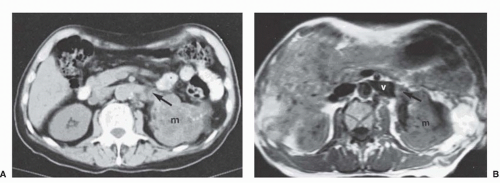
Figure 37.1 Cross-sectional imaging of kidney cancer using computed tomography and magnetic resonance imaging. (A) Contrast-enhanced computed tomography imaging (parenchymal phase) reveals a left renal mass with tumor thrombus within the left renal vein. (B) Magnetic resonance imaging in the same patient shows that the renal vein thrombus extends within the renal vein, but not to the confluence with the inferior vena cava (level 0 thrombus). m, mass; v, vein.
Percutaneous renal mass sampling is now being performed with increased regularity at many centers.29 There is a strong rationale for biopsy when the findings will change management, such as when there is reason to suspect lymphoma/leukemia or abscess or to guide systemic therapy for metastatic disease. Even for clinically localized renal tumors, conventional renal mass biopsy can provide a definitive diagnosis in 80% to 90% of cases, and the ability to subtype and grade RCC can increase with the use of immunohistochemical and other molecular analyses.30,31 Therefore, renal mass sampling should be considered in patients with enhancing renal masses who are candidates for a wide range of management strategies.30,31,32,33,34 However, younger, healthier patients who are unwilling to accept the uncertainty associated with renal mass biopsy as well as elderly, frail patients who will be managed conservatively (independent of biopsy results) should still be managed without a biopsy.
Clinical and pathologic staging systems provide a basis of standardized communication, comparison, and prognostication. They are used to communicate risk for treatment decision making and clinical trials planning. There have been several staging systems for RCC proposed. The most widely used is version 7 of the TNM (tumor, node, metastasis) staging system of the American Joint Committee on Cancer and the Union for International Cancer Control, updated in 2010 (Table 37.2). It distinguishes T1a from T1b and T2a from T2b based on tumor size.35 Additionally, adrenal involvement was changed from pathologic stage T3a to T4 and venous invasion was separated into renal vein/segmental branches (T3a) and inferior vena cava (IVC) below (T3b) or above the diaphragm (T3c). Nodal and metastatic disease are only classified as negative (N0/M0) or positive (N1/M1). Other potential prognostic features of the primary tumor not included in the TNM classification include necrosis, urothelial involvement, microvascular invasion and molecular features. Review of these and other prognostic features of RCC are available.36
HEREDITARY KIDNEY CANCER SYNDROMES, GENETICS, AND MOLECULAR BIOLOGY
While most renal cancers are believed to occur sporadically, familial clusters have led to the discovery of at least seven RCC susceptible syndromes (Table 37.3). It is estimated that approximately 4% of RCC have a hereditary basis.37 In these cases, in addition to a provocative family history, tumors tend to be bilateral, multifocal, and arise at an early age of onset. Importantly, the study of hereditary kidney cancer has dramatically changed our understanding of the genetic and molecular basis of RCC and has led to the development of effective, approved therapeutic agents as similar cytogenetic and molecular alterations appear to be shared between sporadic and hereditary RCC (see Fig. 37.2).38
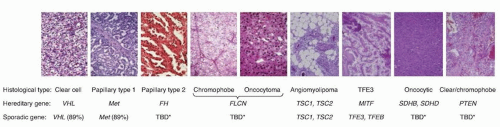
Figure 37.2 Human renal epithelial neoplasms. Renal cortical tumors do not conform to a single pathology. There are a number of different tumor subtypes that display the full range of oncologic activity, ranging from benign to indolent to aggressive. Each histologic type is characterized by distinct gross and microscopic appearance, gene associated with their familial forms, and genetic changes commonly detected in sporadic cases. (Used with permission from Linehan WM, Ricketts CJ. The metabolic basis of kidney cancer. Semin Cancer Biol 2013;23:46-55.)
TABLE 37.2 International Tumor, Node, Metastasis Staging System for Renal Cell Carcinoma and Survival Rates
T: Primary Tumor
Five-Year Survival (%)
TX
Primary tumor cannot be assessed
T0
No evidence of primary tumor
T1a
Tumor ≤4 cm and confined to the kidney
90-100
T1b
Tumor >4 cm and ≤7 cm and confined to the kidney
80-90
T2a
Tumor >7 cm and ≤10 cm and confined to the kidney
65-80
T2b
Tumor >10 cm and confined to the kidney
50-70
T3a
Tumor grossly extends into the renal vein or its segmental (muscle containing) branches, or tumor invades perirenal and/or renal sinus fat but not beyond Gerota fascia
40-65
T3b
Tumor grossly extends into the vena cava below the diaphragm
30-50
T3c
Tumor grossly extends into the vena cava above the diaphragm or invades the wall of the vena cava
20-40
T4
Tumor invades beyond Gerota fascia (including contiguous extension into the ipsilateral adrenal gland)
0-20
N: Regional Lymph Nodes
NX
Regional lymph nodes cannot be assessed
N0
No regional lymph nodes metastasis
N1
Metastasis in regional lymph node(s)
0-20
M: Distant Metastases
MX
Distant metastasis cannot be assessed
M0
No distant metastasis
M1
Distant metastasis present
0-10
Stage Grouping
Stage I
T1
N0
M0
Stage II
T2
N0
M0
Stage III
T3
Any N
M0
T1 or T2
N1
M0
Stage IV
T4
Any N
M0
Any T
Any N
M1
Modified from American Joint Committee on Cancer. Edge S, Byrd DR, Compton CC, et al, eds. AJCC Cancer Staging Manual. 7th ed. New York: Springer-Verlag; 2010. Data from Hafez KS, Fergany AF, Novick AC. Nephron sparing surgery for localized renal cell carcinoma: impact of tumor size on patient survival, tumor recurrence, and TNM staging. J Urol 1999;162(6): 1930-1933; Leibovich BC, Cheville JC, Lohse CM, et al. Cancer specific survival for patients with pT3 renal cell carcinoma—can the 2002 primary tumor classification be improved? J Urol 2005;173(3):716-719; Thompson RH, Cheville JC, Lohse CM, et al. Reclassification of patients with pT3 and pT4 renal cell carcinoma improves prognostic accuracy. Cancer 2005;104:53-60; Lane BR, Kattan MW. Prognostic models and algorithms in renal cell carcinoma. Urol Clin North Am 2008;35:613-625; Campbell L, Nuttall R, Griffiths D, et al. Activated extracellular signal-regulated kinase is an independent prognostic factor in clinically confined renal cell carcinoma. Cancer 2009; 115:3457-3467; Martinez-Salamanca JI, Huang WC, Millán I, et al; International Renal Cell Carcinoma-Venous Thrombus Consortium. Prognostic impact of the 2009 UICC/AJCC TNM staging system for renal cell carcinoma with venous extension. Eur Urol 2011;59:120-127; Haddad H, Rini BL. Current treatment considerations in metastatic renal cell carcinoma. Curr Treat Options Oncol 2012;13:212-229.
The molecular alterations in RCC appear to converge on similar pathways involved in dysregulated oxygen sensing/angiogenesis, iron metabolism, and energy/nutrient sensing.39,40 The predominant genetic and molecular defects in RCC known to date include VHL loss of function (ccRCC), neurofibromatosis type 2 loss of function (ccRCC), MET gain of function (papillary type I RCC), NRF2 gain of function and alterations in fumarate hydratase (papillary type II RCC), CCND1 gain of function and folliculin loss of function (oncocytoma/chromophobe RCC), and MiTF-TFE3 gain of function (translocation RCC). Additionally, inactivation of chromatin modifying proteins including PBRM1, BAP1, and histone methylases/demethylases, as well as inactivation of electron transporters, may represent early or common events in multiple subtypes.41 While important, the associations of these aberrant pathways with various subtypes of RCC likely represent an overly simplified model of renal tumor development.42 A variety of small nucleotide mutations, structural mutations, and large chromosomal abnormalities characterizes RCC, with as many as 5 to 70 small somatic mutations found in individual renal tumor cells.42 Moreover, variable gene expressions may reflect differences in cell types from which RCC originates, suggesting that genetic aberrations require a specific cellular context for dysregulated growth. The development of rapid sequencers continues to redefine the molecular characterization of RCC from which a genetic profile/classification is emerging with important implications for the development of the next generation of targeted therapeutic molecules.43,44
Von Hippel-Lindau Disease
VHL is a syndrome characterized by the development of highly vascular tumors of the retina, central nervous system, pancreas, adrenal, and kidney (ccRCC). It is inherited in an autosomal dominant fashion with an incidence of 1:35,000.39 Loss of VHL function (3p25.1) by genetic or epigenetic means in a classic tumor suppressor fashion is the known cause. In an established genotype-phenotype relationship, type 1 VHL (absence of pheochromocytoma) is due to germline deletions, insertions, and nonsense mutations, whereas type II VHL (with pheochromocytoma) is associated with missense mutations.45 Between 25% to 60% of patients with VHL develop bilateral multifocal cystic and solid RCC, which represents a common cause of death (Fig. 37.3). Management of renal tumors in patients with VHL now includes surveillance of smaller tumors (<3 cm) and resection of larger ones (>3 cm) by PN with the goal of preventing metastases and optimizing renal function by “resetting the biologic clock” through appropriately timed surgeries.46 The goal of complete tumor removal with wide negative surgical margins is less appropriate for these patients where management of localized lesions supplants cure.39,46 Patients should be evaluated and followed by a team of clinicians familiar with the complexities of multisystem genetic disorders.
Hereditary Papillary Renal Cell Carcinoma
Hereditary papillary RCC is perhaps the least common familial RCC syndrome, with manifestations that appear to only affect the kidney. Affected individuals develop bilateral multifocal type I papillary RCC due to mutations in the MET gene located at 7q31. MET is a tyrosine kinase receptor with hepatocyte growth factor as its ligand.47 The syndrome is transmitted in an autosomal dominant fashion and tumors usually appear after the age of 30.39 As with VHL, management of renal tumors recognizes the need to remove larger lesions and observe smaller ones. While no size cutoff for intervention has been established, the biology of type I papillary RCC appears to be more indolent that ccRCC, suggesting the risk of death from kidney cancer in these patients is low. Again, PN with renal preservation is emphasized despite the diffuse micro- and macromultifocality of these lesions.
Succinate dehydrogenase complex subunits: SDHB (1p36.1-35) or SDHD (11q23)
Chromophobe, clear cell, type 2 papillary RCC, oncocytoma
Paragangliomas (benign and malignant)
Papillary thyroid carcinoma
Tuberous sclerosis
TSC1 (9q34) or TSC2 (16p13)
Multiple renal angiomyolipomas
Clear cell RCC (occasionally)
Renal cysts/polycystic kidney disease
Cutaneous angiofibromas
Pulmonary lymphangiomyomatosis
PTEN hamartoma tumor syndrome (Cowden syndrome)
PTEN (10q23)
Breast tumors (malignant and benign)
Epithelial thyroid carcinoma
Papillary RCC or other histology
PTEN, phosphatase and tensin homolog; RCC, renal cell carcinoma.
Adapted from Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol 2003; 170(6 Pt 1):2163-2172. and Linehan WM, Ricketts CJ. The metabolic basis of kidney cancer. Semin Cancer Biol 2013;23:46-55.
Birt-Hogg-Dubé Syndrome
BHD is characterized by cutaneous fibrofolliculomas, a 50-fold increased risk of pneumothorax, and bilateral multifocal solid renal tumors. It is an autosomal dominant disorder with an incidence of around 1:200,000. Linkage analysis has mapped the gene for BHD (folliculin), a tumor suppressor, to 17p11.2. Folliculin is thought to be a downstream effector of activated protein kinase and mammalian target of rapamycin (mTOR).39 Renal tumors associated with BHD are more indolent in nature, occurring in approximately 20% of individuals, with <5% developing metastases. While the histology of renal tumors associated with BHD is most often chromophobe RCC, oncocytoma, or hybrids of both, clear cell or papillary tumors may rarely occur as well.
Hereditary Leiomyomatosis Renal Cell Carcinoma
Hereditary leiomyomatosis RCC is also characterized by dermatologic manifestations. Patients with HLRCC exhibit cutaneous leiomyomas, early onset of uterine fibroids, macronodular adrenal hyperplasia, and kidney cancer. Linkage analysis has localized the HLRCC gene (fumurate hydratase), a key Kreb cycle enzyme, to 1p42.3 (FN).48 Renal tumors in HLRCC tend to be aggressive and lethal. The most common histology observed is type II (eosinophilic) papillary RCC.39 Unlike other hereditary forms of RCC, given the aggressive nature of these tumors, AS with delayed intervention is not recommended.
TREATMENT OF LOCALIZED RENAL CELL CARCINOMA
Greater use of cross-sectional imaging has contributed to earlier detection of RCC in many cases.2,49,50 Between 50% to 70% of RCC are detected incidentally,51 and the majority are “small renal masses (SRM),” or clinically localized renal cortical tumors up to 4 cm in size. Our perspectives about treatment of clinical T1 renal masses have changed substantially in the past 20 years. While all had been presumed to be malignant and managed aggressively, we now recognize the tremendous biologic heterogeneity of these lesions, and multiple management strategies are now available, including RN, PN, TA, and AS.52,53,54,55,56,57 Once controversial, elective PN is now accepted as a standard of care, based in part on the appreciation of the deleterious renal functional consequences of RN (Fig. 37.4).53,55,58 Ongoing analysis of the relative merits of PN, RN, and other management strategies has produced vibrant literature over the past few years.46,59,60,61,62 Both the American Urologic Association (AUA) and the European Association of Urology have released guidelines for the management of localized renal masses in recent years providing a robust analysis of the available studies.9,63,64,65,66,67 Each approach has associated risks and benefits, and no one approach is best in all circumstances (Table 37.4). The involvement of an urologist with expertise in the management of RCC is essential for selection of the optimal strategy based on the individual features of each patient and tumor.
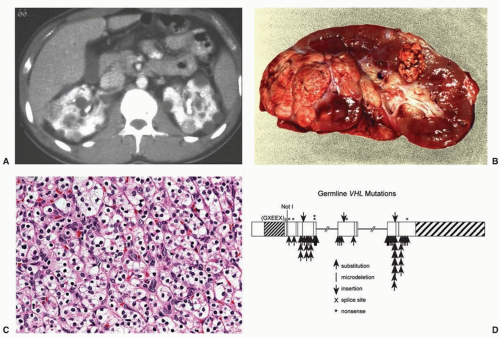
Figure 37.3 The von Hippel-Lindau (VHL) gene is responsible for the inherited form of clear cell renal cell carcinoma (ccRCC): VHL syndrome. (A) Axial computed tomography image showing multifocal and bilateral renal tumors and cysts. (B) Gross image of nephrectomy specimen showing typical yellow-gold appearance of ccRCC present in multiple portions of this kidney from a patient with VHL. (C) Histologic appearance of ccRCC, showing the clearing of the cytoplasm around the darker nuclei typical of these “clear cells.” (D) Structure of the VHL gene with sites of point mutations and truncations indicated. (Used with permission from Linehan and colleagues’ prior work. DeVita VT, Lawrence TS, Rosenberg SA, et al, eds. DeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.)
Radical Nephrectomy for Renal Cell Carcinoma
The objective of surgical therapy for RCC is to excise all of the cancer with an adequate surgical margin. Simple nephrectomy was practiced for many decades, but was replaced by RN when Robson and colleagues (1969) established this procedure as the “gold standard” approach for localized RCC.68 “RN” as currently practiced may be better termed “total nephrectomy,” as it often omits several of the components of the original, “radical” nephrectomy, which always included extrafascial nephrectomy, adrenalectomy, and extended lymphadenectomy from the crus of the diaphragm to the aortic bifurcation. Perifascial dissection is still routinely practiced for larger tumors, as ≥25% of these tumors extend into the perinephric fat.69,70 Removal of the ipsilateral adrenal gland is no longer recommended, unless there is suspicion of direct invasion of the gland by tumor or a radiographically or clinically suspicious adrenal tumor, because of the similar propensity of RCC to metastasize to the ipsilateral or contralateral adrenal gland.71,72,73 Finally, extended lymphadenectomy has been shown to be of no benefit for patients with clinically localized RCC and remains controversial for those with higher-risk disease.74,75,76,77
RN is still a preferred option for many patients with localized RCC, such as those with very large tumors (most clinical T2 tumors) or the relatively limited subgroup of patients with clinical T1 tumors that are not amenable to nephron-sparing approaches.78 The surgical approach for RN depends on the size and location of the tumor, as well as the patient’s habitus and medical/surgical history. For locally advanced disease and/or bulky lymphadenopathy, an open surgical approach using either an extended subcostal, midline, or thoracoabdominal incision is generally used.79
Current minimally invasive approaches allow all of the essential steps of RN to be performed, with the associated benefits of shorter convalescence and reduced morbidity.80,81,82,83 Laparoscopic RN is now established as a preferred approach for moderate to large volume tumors (≤10 cm to 12 cm), without invasion of adjacent organs, with limited (or no) venous involvement, and having manageable (or no) lymphadenopathy. Therefore, a minimally invasive approach is suitable for most patients with renal tumors, even including some patients with features previously thought to mandate open RN.
On the other hand, RN has fallen out of favor for smaller renal tumors due to concerns about chronic kidney disease (CKD), and it should only be performed when necessary in this population.53,58,78,84 Several studies have shown an increased risk of CKD on longitudinal follow-up after RN.58,85,86 Huang et al. first reported that 26% of patient populations with a small renal mass, normal opposite kidney, and “normal” serum creatinine had preexisting grade 3 CKD (estimated glomerular filtration rate [eGFR] <60 ml/min per 1.73 m2). After surgery, stage 3b or higher CKD (eGFR <45 ml/min per 1.73 m2) was more common after RN than PN (36% versus 5%, p <0.001). CKD has been proven to lead to increased rates of cardiovascular events and death, with proportionally greater impact with higher CKD stage (and lower GFR). For example, in a population-based study of >1 million subjects, the relative death rates were 1.2, 1.8, 3.2, and 5.9 for eGFR (ml/min per 1.73 m2) of 45 to 60, 30 to 45, 15 to 30, and <15, respectively, even after controlling for hypertension, diabetes, and other potential confounding factors.87 Coupled with the biologic heterogeneity of small renal masses, many of which will never lead to compromised survival, the potential negative consequences of RN on renal function have highlighted the importance of nephron-sparing approaches.53,58,72,88,89,90
Figure 37.4 Partial nephrectomy. The intention of kidney-sparing surgery, or “nephron-sparing surgery,” is to achieve complete local resection of the tumor while leaving as much functioning parenchyma in the involved kidney as possible. An assessment of volume preservation can be made by accounting for both the amount of parenchyma replaced by tumor and the adjacent uninvolved parenchyma removed or devascularized during the procedure.61 The amount of volume preservation and the quality of the functioning renal remnant are the most important determinants of renal function after renal surgery. Partial nephrectomy and other kidney-sparing alternatives provide definite renal functional benefits that must be weighed against the potential for increased risk of cancer recurrence, when compared with radical nephrectomy.287 (Artwork courtesy of Kristen Tobert.)
Partial Nephrectomy for Renal Cell Carcinoma
Kidney-sparing surgery for renal tumors was first described by Czerny in 1890; however, significant morbidity limited its use for the next half century.91 Vermooten (1950) revisited the concept of local excision with a margin of normal parenchyma for encapsulated and peripherally located renal tumors.92 The use of PN for RCC has subsequently been stimulated by experience with renal vascular surgery for other conditions, advances in renal imaging, growing numbers of incidentally discovered small renal masses, greater appreciation of the deleterious effects of CKD, introduction of minimally invasive techniques, and encouraging long-term survival in patients undergoing this form of treatment during the last 50 years.93,94,95 Kidney-sparing surgery entails complete local resection of the tumor while leaving the largest possible amount of normal functioning parenchyma in the involved kidney (see Fig. 37.4).
Initially described for patients with an “absolute” indication for kidney-sparing surgery or for the “elective” indication of a small renal tumor in the setting of a normal contralateral kidney, PN is now strongly considered whenever preservation of renal function is potentially important. Common indications include conditions that pose a threat to future renal function, such as hypertension, diabetes mellitus, peripheral or coronary artery disease, or nephrolithiasis, and patients with baseline CKD, an abnormal contralateral kidney, or those with multifocal or familial RCC.96,97 PN is generally considered feasible for the vast majority of localized renal masses <5 cm in size and often for tumors ≥7 cm by those with expertise with kidney-sparing surgery.98,99,100,101 Particularly for those with a strong rationale for nephron-sparing surgery, PN can even be performed for tumors that deeply invest the renal vascular structures or with limited venous thrombus, but such procedures clearly carry higher perioperative morbidity.95,102 Local recurrence rates after PN for imperative indications have averaged 3% to 5% or higher in historical series.103 The decision to perform a PN in such circumstances should be individualized, weighing the potential increased technical and oncologic risks of such an operation with the renal functional consequences of RN (see Fig. 37.4).
PN clearly leads to improved functional outcomes, when compared with RN, even for complicated situations.104,105 Temporary or permanent renal replacement therapy has been reported to be necessary in <5% of patients undergoing PN in a solitary kidney and is rarely needed for patients with a functioning contralateral kidney.106,107 In fact, the vast majority of patients will avoid permanent dialysis, even following multiple surgeries for multiple tumors in both kidneys, so long as at least 30% of a well-functioning remnant kidney is preserved.95 For situations in which PN is deemed impossible, RN with ensuing hemodialysis is sometimes necessary, but presurgical therapy with a tyrosine-kinase inhibitor is an alternative approach that has proven successful in some patients.108,109,110,111
For patients with clinical T1 renal masses, local recurrence rates are 1% to 2% after PN and most commonly located distant from the initial resection. Cancer-free survival is achieved in well above 90% of patients.104 The contralateral kidney is also at risk for metachronous disease, which also occurs in 1% to 2% of patients, even with contemporary imaging modalities. This provides further rationale to avoid unnecessary RN for tumors amenable to nephron-sparing surgery. The goal of PN is resection of all grossly appreciated tumor with negative microscopic surgical margins; this is generally performed with a thin rim of normal parenchyma based on prior literature indicating that margin width is immaterial.67,112,113,114,115,116,117 Some centers now routinely perform enucleation of renal tumors with excellent oncologic outcomes, although enthusiasm for more widespread use of this approach has been tempered by the somewhat higher recurrence rate among patients with RCC with positive margins and the propensity of some RCC subtypes to invade the pseudocapsule that is generally present.
Within the last decade, substantial progress has been made with minimally invasive PN, which is now the most commonly performed procedure for small renal masses. Laparoscopic PN, with or without robotic assistance, is performed according to the same principles as open PN. Margin status and oncologic outcomes with laparoscopic and open PN appear equivalent in series of patients in which patients were selected appropriately for each of these approaches.101,118 Although early to intermediate experience with laparoscopic PN suggested increased urologic complications compared with open RN,119 subsequent experience with pure laparoscopic PN and more prevalent use of robotic PN have substantially reduced perioperative morbidity.104,120,121,122,123,124,125 Tumor complexity remains a major predictor for intraoperative and postoperative complications, regardless of surgical approach, and open PN should be considered for particularly challenging situations.126
TABLE 37.4 Treatments for Localized Renal Cell Carcinoma
Advantages
Disadvantages
Main Indications
ORN
Traditional surgical approach for renal cancer, effective in removing tumor with surrounding structures and lymph nodes when indicated
Morbidity of surgical incision (flank, subcostal, midline, thoracoabdominal)
Renal functional implications of removing entire kidney (average 35% decrease in GFR)
Large tumor (>12 cm)
Locally advanced tumor
Bulky adenopathy
MIRN
Reproducible and effective surgery for most localized renal tumors
Minimally invasive surgery, with decreased pain, morbidity and convalescence compared to ORN
Many tumors up to 7 cm can be treated with PN
Renal functional implications of removing entire kidney (average 35% decrease in GFR)
Medium to large tumor (up to 10 to 12 cm)
High tumor complexity
OPN
Oncologic outcomes appear similar to RN, although selection biases limit this conclusion
Maximizes renal functional preservation when performed with precise tumor excision and judicious use of regional hypothermia
Morbidity of flank incision (bulge, longer recovery)
Potential for local recurrence due to incomplete excision or de novo tumors in the renal remnant
Small to medium tumors (up to 7 cm and occasionally larger)
Moderate to high-complexity tumors
MIPN
Kidney-sparing surgery, with preservation of renal function when warm ischemia kept to limited duration (<20 to 25 min)
Minimally invasive surgery, with decreased pain, morbidity, and convalescence compared to OPN
Higher complication rate for high complexity tumors and in lessexperienced hands
Positive surgical margins and local recurrence rates may be higher in such situations
Small renal masses (up to 5 cm and occasionally larger)
Low to moderate (and selected high) complexity tumors
TA
Kidney-sparing approach, with renal functional benefits versus RN
Can be performed outside of OR (percutaneous) or with minimally invasive approach (laparoscopic)
For small (<3 cm) tumors, provides comparable control of metastasis to PN and RN
Relatively high rate of local failure
Imprecision of histopathologic diagnosis
Increased and challenging radiographic follow-up
Prior ipsilateral surgery for renal tumor
Poorer surgical candidates unwilling to undergo surveillance
AS
Least invasive and kidney-sparing of all strategies
Most SRMs have limited oncologic potential and can be safely managed with initial short interval follow-up imaging
Intensity of surveillance can be tailored to patient and tumor characteristics
Tumor remains in place and untreated
Oncologic nature of tumor unknown (without biopsy)
Poor surgical candidates
Limited life expectancy
ORN, open radical nephrectomy; GFR; glomerular filtration rate; MIRN, minimally invasive radical nephrectomy; OPN, open partial nephrectomy; RN, radical nephrectomy; MIPN, minimally invasive partial nephrectomy; TA, thermal ablation; OR, operating room; PN, partial nephrectomy; AS, active surveillance; SRM, small renal mass.
Thermal Ablation of Renal Cell Carcinoma
TA, including renal cryosurgery and radiofrequency ablation (RFA) have emerged as alternative kidney-sparing treatments for patients with small (<3 cm) renal tumors.127,128 Both can be administered percutaneously or with laparoscopic exposure and thus offer the potential for reduced morbidity and more rapid recovery compared with extirpative approaches.128,129 In general, the long-term efficacy of TA has not been well established when compared to surgical excision, and current data suggest that the local recurrence rates are somewhat higher than that reported with extirpative approaches.55,63 Another concern has been the lack of accurate histologic and pathologic information obtained with these modalities because the treated lesion is left in situ.
The ideal candidates for TA are patients with advanced age and/or significant comorbidities who prefer a proactive approach (over surveillance), but are not optimal candidates for conventional surgery, patients with local recurrence after previous kidney-sparing surgery, and patients with hereditary renal cancer who present with multifocal lesions for which multiple PNs might be cumbersome.55 Patient preference must also be considered as some patients not fitting these criteria may select TA after balanced counseling about the current status of these modalities.130,131 Tumor size is an important factor in patient selection because the current technology does not allow for reliable treatment of lesions >4 cm and success rates appear to be highest for tumors <2.5 cm to 3 cm.132,133,134,135
Clinical experience and follow-up of patients after renal cryosurgery suggests successful local control in about 90% of patients, although many studies provide limited and often incomplete, follow-up.136,137,138,139,140 Diagnosis of local recurrence after TA can be challenging because evolving fibrosis within the tumor bed can be difficult to differentiate from residual cancer. In general, central or nodular enhancement within the tumor bed on extended follow-up has been considered diagnostic of local recurrence and the clinical experience with cryoablation has thus far supported this.141,142 Other findings that suggest local recurrence include failure of the treated lesion to regress over time, a progressive increase in size of an ablated neoplasm, new nodularity in or around the treated zone, or satellite or port site lesions.143 If these features are found, biopsy and possible retreatment should be considered. The AUA guidelines for surveillance after TA include cross-sectional scanning (computed tomography or MRI) with and without intravenous contrast at 3 and 6 months following ablative therapy and annually with chest X-ray for 5 years thereafter.143
Only gold members can continue reading. Log In or Register to continue